Clear Sky Science · it
Apprendere dinamiche molecolari coarse-grained data-efficienti da forze e rumore
Perché ridurre le molecole è importante
Simulare il movimento incessante di ogni atomo in una proteina e nell’acqua circostante è uno dei nostri migliori strumenti per comprendere come funziona la vita su scala molecolare. Ma queste simulazioni a livello atomico sono così esigenti dal punto di vista computazionale che seguire una proteina mentre si ripiega, si apre o interagisce con partner per tempi rilevanti biologicamente può richiedere mesi su un supercomputer. Questo articolo presenta un nuovo modo di costruire modelli veloci e semplificati delle proteine che si comportano ancora come i loro equivalenti atomici completi, pur richiedendo molti meno dati di addestramento e potenza di calcolo rispetto al passato.
Dall’atomo a un quadro più semplice
La dinamica molecolare tradizionale traccia ogni atomo e calcola le forze tra di essi ad ogni minuscolo passo di integrazione. Per accelerare i calcoli, gli scienziati spesso usano modelli coarse-grained, che raggruppano molti atomi in un numero inferiore di “perle”. Questi modelli ridotti sono molto più veloci ma storicamente hanno faticato a eguagliare l’accuratezza delle simulazioni atomistiche complete, soprattutto per proteine con comportamenti di ripiegamento complessi. Lavori recenti si sono rivolti al machine learning per scoprire automaticamente campi di forza coarse-grained migliori, ma l’addestramento di questi modelli ha tipicamente richiesto milioni di snapshot dettagliati, ognuno etichettato con le forze su ogni atomo—un onere enorme in termini di dati e calcolo.
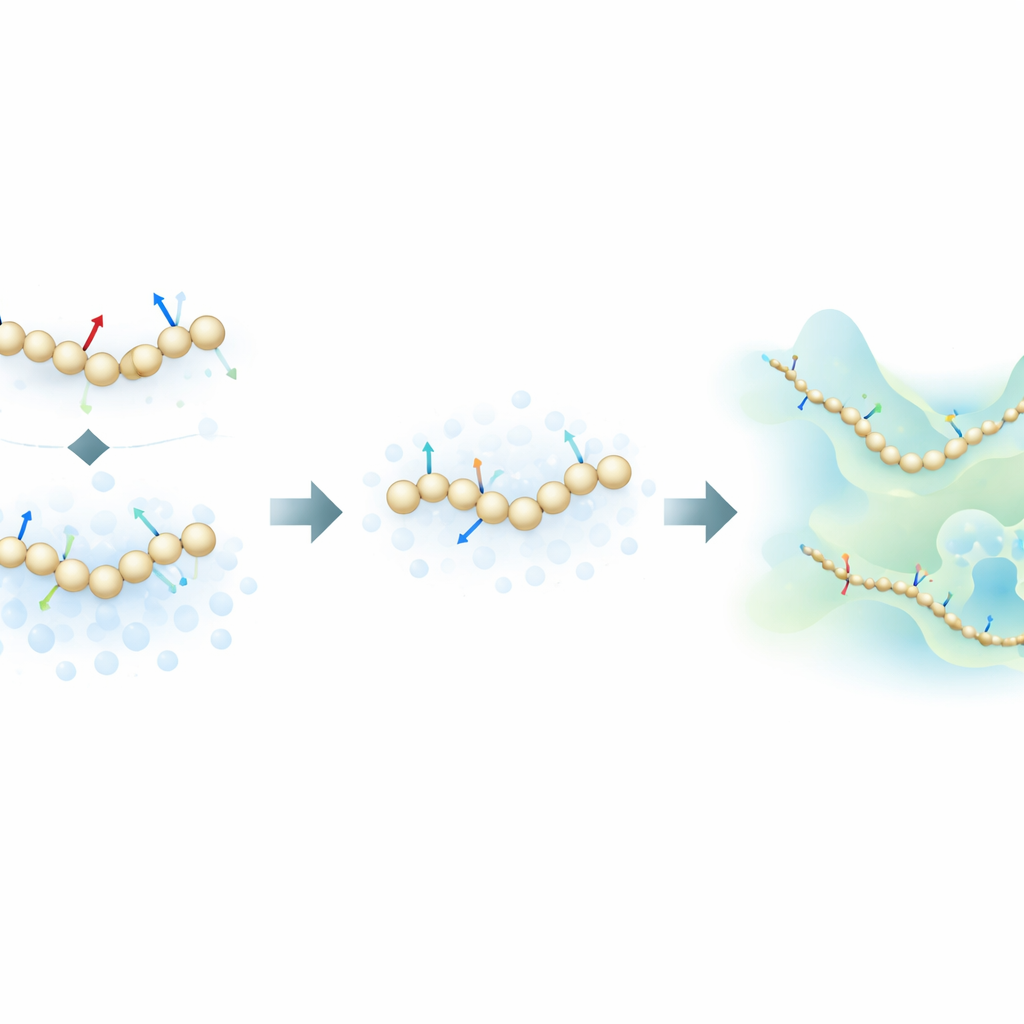
Combinare forze fisiche e rumore informativo
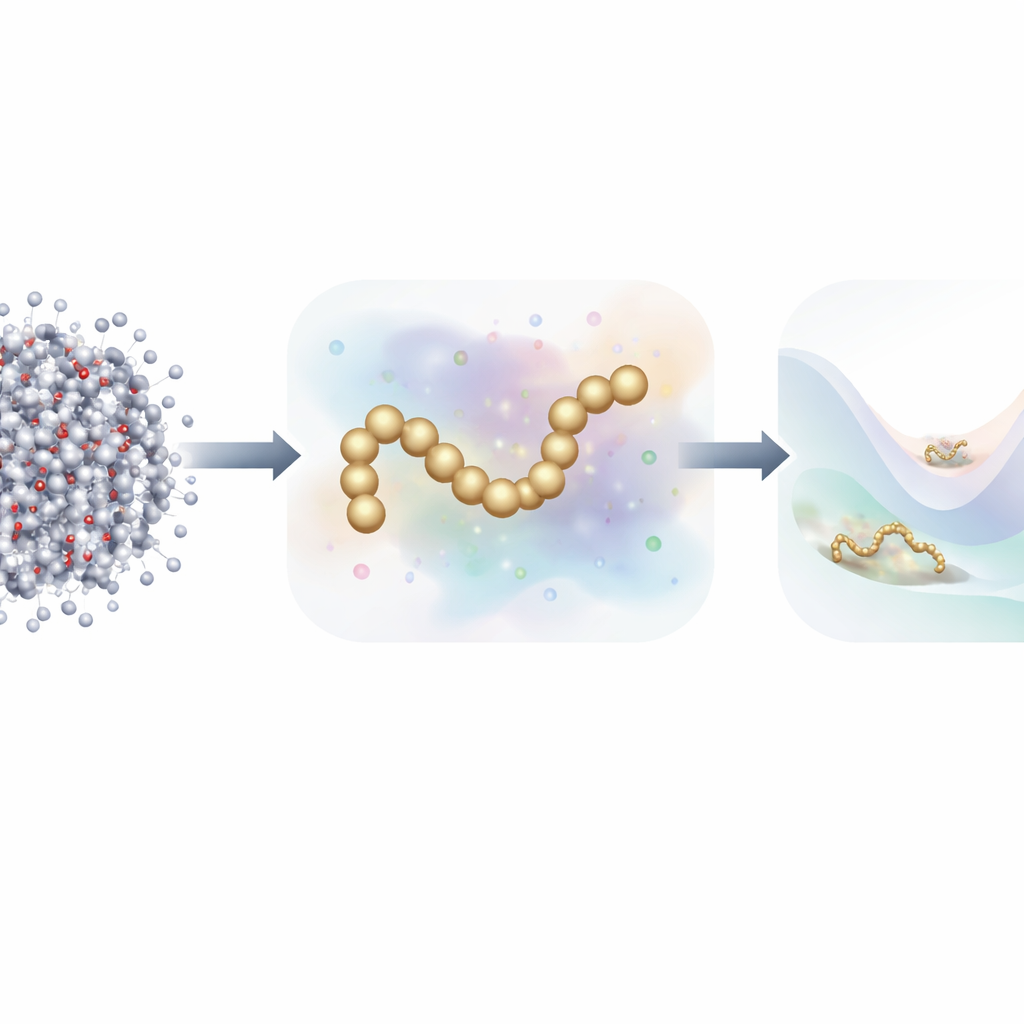
Gli autori propongono una nuova strategia di addestramento che prende ispirazione dai modelli di diffusione generativa—la stessa classe di algoritmi alla base di molti moderni generatori di immagini AI. Invece di apprendere soltanto dalle forze fisiche calcolate nelle simulazioni atomistiche, il loro metodo apprende anche da come le strutture molecolari si distribuiscono nello spazio aggiungendo deliberatamente rumore controllato alle configurazioni coarse-grained. In questo quadro, il rumore non è solo un fastidio da rimuovere; diventa una fonte aggiuntiva di informazioni. Unificando matematicamente il tradizionale approccio di “force matching” con le tecniche di denoising dei modelli di diffusione, il metodo può dedurre il paesaggio energetico sottostante di una proteina usando un numero di esempi etichettati molto inferiore.
Insegnare a modelli semplici a imitare proteine complesse
Per mettere alla prova l’idea, i ricercatori hanno addestrato modelli coarse-grained basati su reti neurali per diverse proteine di complessità crescente: le piccole miniproteine Chignolin e Trp-Cage, la più grande NTL9 e la proteina di 76 residui Ubiquitina. Hanno confrontato tre modalità di addestramento: usando solo le forze atomistiche, usando solo informazioni derivate dal rumore e combinando entrambe. Per le proteine più piccole hanno mostrato che il nuovo approccio combinato può riprodurre le caratteristiche chiave del paesaggio di ripiegamento—come la stabilità relativa degli stati ripiegati e non ripiegati e la presenza di intermedi—usando fino a cento volte meno dati di addestramento rispetto ai metodi standard di force-matching. Sorprendentemente, in regimi con pochi dati, anche i modelli addestrati solo con informazioni da rumore spesso uguagliavano o superavano l’accuratezza dell’addestramento basato solo sulle forze.
Affrontare sistemi proteici più grandi e difficili
Ubiquitina è una prova più impegnativa: catturare il suo ripiegamento e dispiegamento a temperature realistiche ha storicamente richiesto hardware specializzato e corse atomistiche estremamente lunghe. Qui, gli autori addestrano modelli coarse-grained usando un dataset modesto costituito da brevi simulazioni di equilibrio attorno allo stato ripiegato più simulazioni non di equilibrio “tirate” che allungano forzatamente la proteina. Nonostante questo set di addestramento sbilanciato e la mancanza di un riferimento atomistico perfetto alle stesse condizioni, il modello addestrato con forze e rumore recupera un quadro realistico in cui stati ripiegati e non ripiegati coesistono, con lo stato ripiegato favorito in termini di stabilità. Al contrario, un modello addestrato solo sulle forze non riesce affatto a stabilizzare lo stato ripiegato, mentre un modello basato solo sul rumore preferisce strutture dispiegate. È notevole che nessuno dei modelli coarse-grained memorizzi semplicemente le forme estremamente allungate presenti nei dati di addestramento, indicando che il paesaggio energetico appreso è fisicamente significativo e non solo un’impronta delle traiettorie di input.
Cosa significa per le simulazioni future
Trasformando il rumore in un segnale di addestramento e fondendolo con le forze fisiche, questo lavoro mostra che è possibile costruire modelli coarse-grained accurati delle proteine a partire da dataset molto più piccoli e meno ideali di quanto si pensasse in precedenza. In pratica, ciò significa che i ricercatori potrebbero non avere più bisogno di simulazioni atomistiche dell’ordine del millisecondo su supercomputer specializzati prima di poter esplorare il comportamento di una biomolecola con dinamiche coarse-grained apprese via machine learning. Al contrario, simulazioni più modeste su hardware ampiamente disponibile potrebbero essere sufficienti per addestrare modelli ridotti potenti che catturano le principali vie di ripiegamento e gli equilibri termodinamici. Pur rimanendo domande aperte su come scegliere e interpretare al meglio il rumore aggiunto e su come il metodo si comporterà su assemblaggi biomolecolari ancora più grandi e complessi, questo approccio abbassa sostanzialmente la barriera all’uso di simulazioni coarse-grained guidate dai dati come strumento di routine nelle scienze molecolari.
Citazione: Durumeric, A.E.P., Chen, Y., Pasos-Trejo, A.S. et al. Learning data-efficient coarse-grained molecular dynamics from forces and noise. Nat Commun 17, 2493 (2026). https://doi.org/10.1038/s41467-026-70818-0
Parole chiave: dynamiche molecolari coarse-grained, campi di forza basati su machine learning, simulazioni del ripiegamento delle proteine, modelli di diffusione in chimica, simulazione data-efficient