Clear Sky Science · it
Una base metrologica per la trascrittomica assoluta usando calibratori ancorati al Sistema Internazionale di unità
Perché trasformare i segnali dell'RNA in numeri reali è importante
I test genetici moderni possono leggere quali geni sono attivati o disattivati nelle nostre cellule, ma inciampano su una domanda fondamentale: quante molecole sono davvero presenti? Le tecnologie di sequenziamento dell'RNA attuali confrontano per lo più cambiamenti relativi tra campioni invece di fornire conteggi assoluti e affidabili. Questo è un problema se si vogliono definire soglie universali per le malattie, confrontare risultati tra ospedali o costruire modelli precisi del funzionamento cellulare. Questo studio introduce un nuovo modo di ancorare il sequenziamento dell'RNA alle stesse unità internazionali usate in chimica e fisica, trasformando segnali relativi sfumati in numeri assoluti e confrontabili.
Il problema del confronto dell'attività genica
Il sequenziamento dell'RNA funziona frammentando le molecole di RNA e contando quante volte ogni gene è rappresentato. Ma si insinuano due tipi di distorsione. Primo, differenze sistematiche tra esperimenti – come diversi laboratori, macchine o metodi di preparazione dei campioni – creano “effetti batch” che fanno apparire diverso lo stesso campione se elaborato più volte. Secondo, effetti dipendenti dalla sequenza – dove geni con certe lunghezze o composizioni in basi sono più o meno propensi a essere catturati – significano che anche all'interno di un singolo campione alcuni geni sono costantemente sovracontati e altri sottocontati. Di conseguenza, gli scienziati sono in gran parte costretti a parlare di rapporti di variazione tra condizioni invece dei veri conteggi di molecole, e anche questi rapporti possono essere fuorvianti da un batch all'altro.
Un nuovo set di riferimenti per le misure di RNA
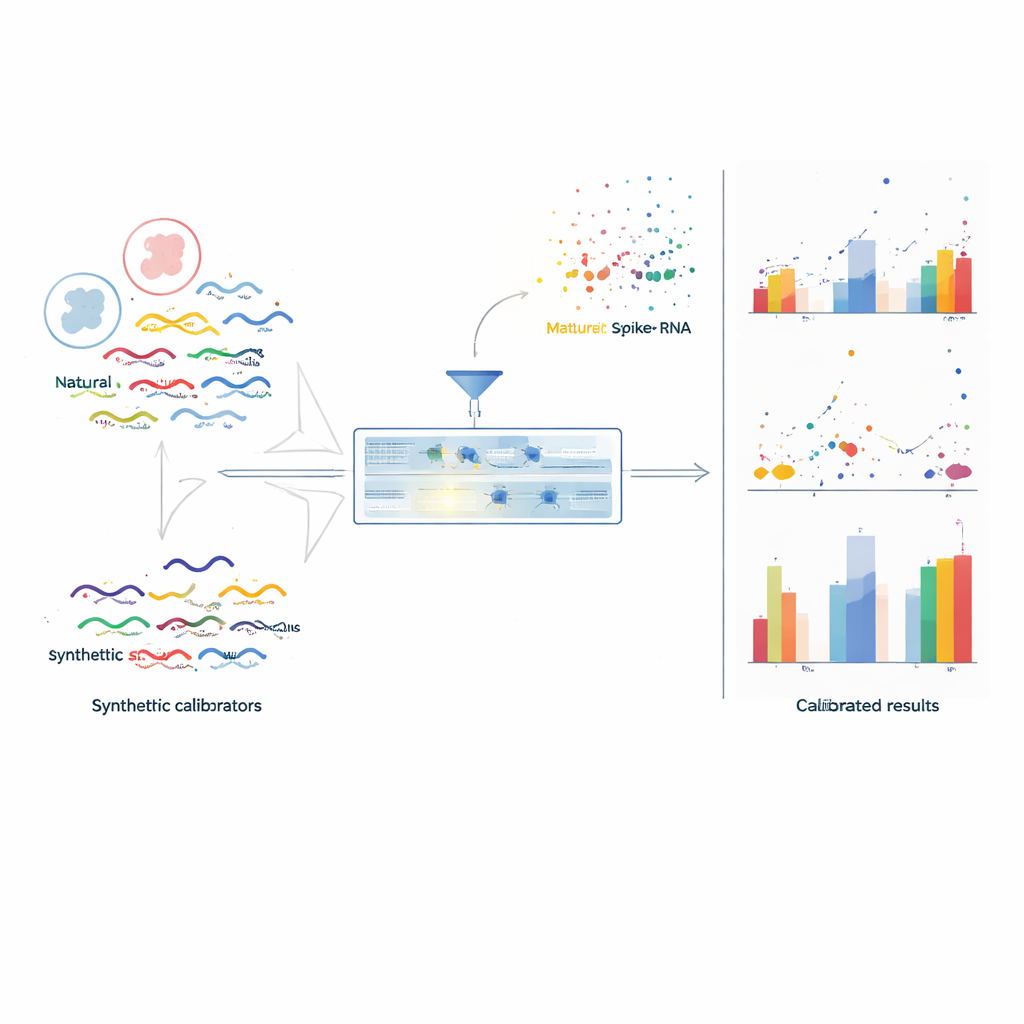
Per risolvere il problema, gli autori hanno creato TranScale, un pannello di 100 molecole sintetiche di RNA progettate per comportarsi come veri trascritti umani pur rimanendo distinti a livello computazionale. Questi standard coprono un ampio intervallo di lunghezze, caratteristiche di sequenza e varianti clinicamente rilevanti come isoforme di splicing e fusioni geniche, rispecchiando da vicino la diversità dell'RNA cellulare reale. Fondamentale, a ogni molecola TranScale è stata assegnata una concentrazione esatta usando una tecnica di misura primaria chiamata spettrometria di massa con diluizione isotopica, tracciabile al Sistema Internazionale di unità (SI). Mischiando una quantità nota e piccolissima di TranScale in ogni campione di RNA prima del sequenziamento, l'esperimento acquisisce un righello interno che attraversa gli stessi passaggi di laboratorio e subisce le stesse distorsioni degli RNA naturali.
Trasformare letture rumorose in conteggi assoluti
Con TranScale presente in ogni libreria, il team può confrontare il numero di letture di sequenziamento per ciascuna molecola spike-in con la sua concentrazione certificata. Per ogni batch selezionano spike-in ben comportati e adattano una curva di calibrazione lineare che collega le unità basate sulle letture ai veri conteggi di molecole. Questo modello semplice cattura simultaneamente sia i bias a livello di batch sia quelli legati alla sequenza. La stessa curva viene poi applicata a tutti i geni nel campione, convertendo i loro segnali relativi in numeri assoluti di copie per unità di RNA. In uno studio su larga scala multi-laboratorio e multipiattaforma progettato appositamente per produrre forti effetti batch, questa calibrazione ha ridotto la variazione mediana delle misure assolute tra centri da oltre l'85% a meno del 15–25%, e ha ripristinato il corretto raggruppamento dei campioni biologici che era stato oscurato dal rumore tecnico.
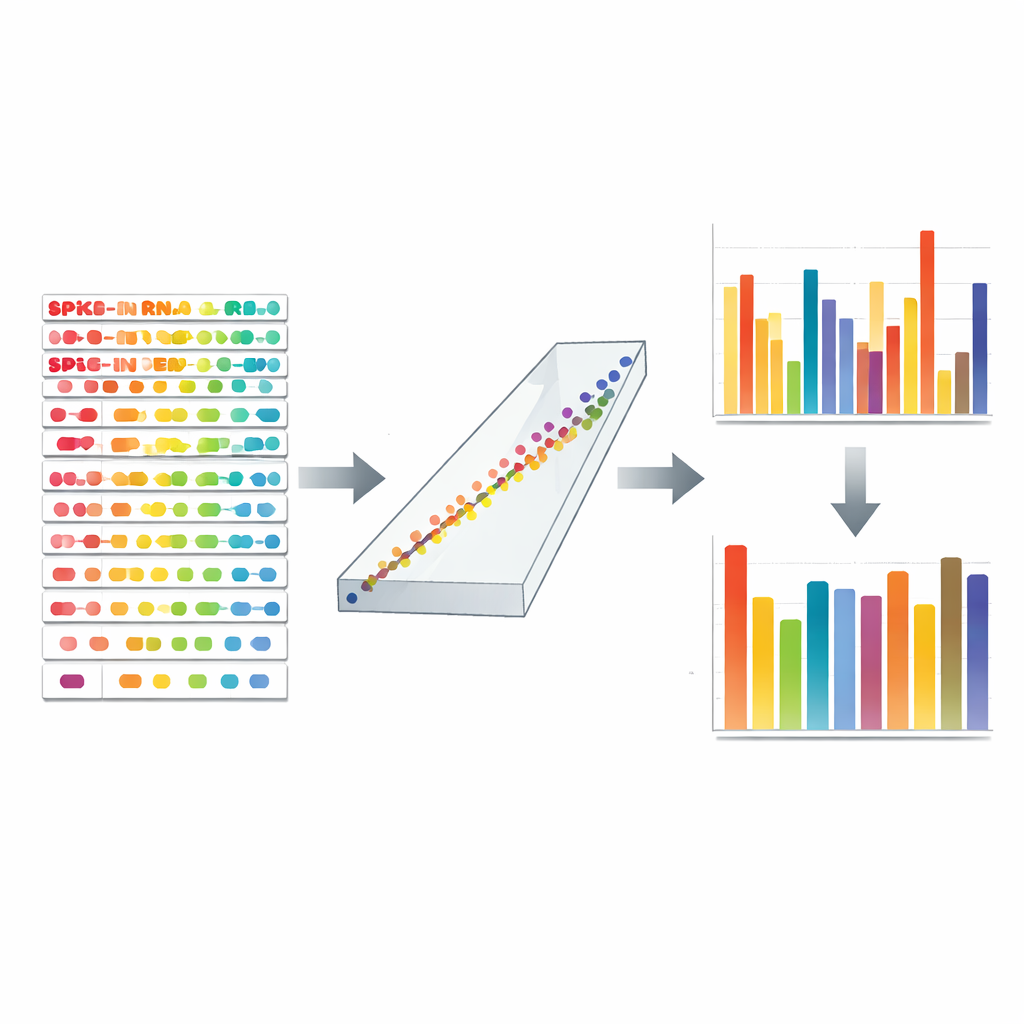
Individuare errori nascosti e correggerli
Gli standard TranScale fungono anche da sonde diagnostiche della qualità dei dati. Confrontando i valori misurati con le loro verità certificate, gli autori hanno separato due tipi di errore: quanto è sbagliato il livello assoluto di ciascun gene e quanto sono errati i rapporti tra condizioni. Hanno trovato esempi sorprendenti in cui le differenze relative sembravano coerenti ma i numeri assoluti erano gravemente distorti, e viceversa. Ciò significa che i controlli convenzionali che si concentrano solo sui rapporti di variazione possono perdere problemi seri. Dopo la calibrazione, sia i livelli assoluti sia i rapporti degli spike-in e di migliaia di geni umani reali corrispondevano strettamente a misure indipendenti di PCR digitale e a un dataset di riferimento esterno. I dati corretti hanno rivelato un panorama quantitativo molto più nitido, rendendo possibile confrontare geni housekeeping e oncogeni sulla stessa scala assoluta e collegare direttamente le variazioni del DNA, come geni oncogeni co-amplificati, alle loro uscite in RNA.
Dalle tendenze relative alle soglie cliniche
Infine, i ricercatori hanno mostrato come la scala assoluta possa affinare le decisioni cliniche. Usando un oncogene spesso misurato nel cancro al seno, hanno definito una soglia fissa basata su PCR digitale e verificato se il sequenziamento dell'RNA potesse classificare in modo affidabile campioni come normali o tumorali attraverso molti batch. I dati non corretti fornivano risposte incoerenti a causa degli effetti batch. Dopo la calibrazione con TranScale, ogni libreria concordava con la classificazione vera. Legando il sequenziamento dell'RNA alle unità SI tramite standard biomimetici, questo lavoro pone una base metrologica per la trascrittomica. Apre la strada a soglie diagnostiche universali, a una condivisione dei dati robusta tra centri e a modelli più precisi e a livello di sistema su come i geni vengono espressi in salute e malattia.
Citazione: Zhang, Y., Yang, B., Yu, Y. et al. A metrological foundation for absolute transcriptomics using International System of Units-anchored calibrators. Nat Commun 17, 2747 (2026). https://doi.org/10.1038/s41467-026-70582-1
Parole chiave: sequenziamento dell'RNA, quantificazione assoluta, metrologia, calibrazione dell'espressione genica, standard biomolecolari