Clear Sky Science · it
Una mutazione patogena di Tau causa disfunzione autofagia‑lisosoma che limita la degradazione di Tau in un modello di demenza frontotemporale
Quando le squadre di pulizia del cervello restano indietro
Perché alcune persone sviluppano gravi problemi di memoria e comportamento decenni prima della vecchiaia? Questo studio affronta la domanda concentrandosi su una singola proteina cerebrale, Tau, e sui minuscoli «centri di riciclaggio» cellulari che normalmente la mantengono sotto controllo. Osservando neuroni umani viventi con microscopi ad altissima risoluzione, i ricercatori mostrano come una mutazione di Tau che causa la malattia intasi il sistema di smaltimento dei rifiuti della cellula e come stimolare quel sistema con una piccola molecola possa aiutare a sgomberare il disordine. I risultati potrebbero indicare nuove strategie terapeutiche per certe forme di demenza.
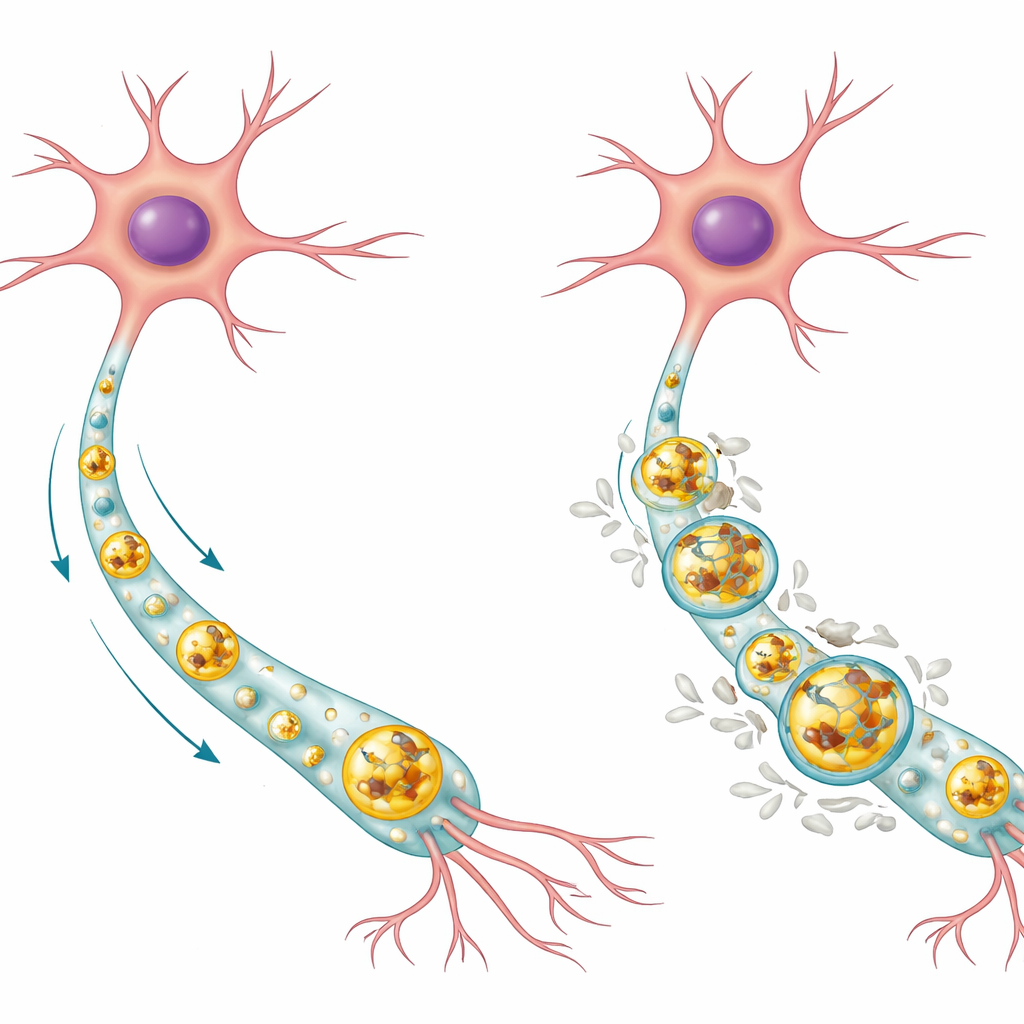
Come i neuroni normalmente portano fuori la spazzatura
I neuroni sono cellule a lunga durata che non possono semplicemente dividersi per diluire il materiale danneggiato, quindi fanno molto affidamento sui sistemi di pulizia interni. Una via chiave è il percorso autofagia‑lisosoma. In questo processo, proteine indesiderate e parti usurate vengono avvolte in sacche membrane‑ri chiamate autofagosomi, che poi si fondono con compartimenti pieni di enzimi noti come lisosomi, dove il carico viene degradato e riciclato. Nei neuroni umani sani, gli autori hanno osservato che la Tau normale tende ad accumularsi all’interno del centro acido dei lisosomi, dove può essere degradata, mentre la forma fosforilata di Tau (una modifica chimica legata alla malattia) si trova più spesso sulla membrana esterna del lisosoma. La maggior parte dei lisosomi nelle cellule sane risultava del tutto priva di Tau, suggerendo che questo sistema di solito mantiene i livelli di Tau bassi e ben controllati.
Cosa va storto in una forma genetica di demenza
Il gruppo si è concentrato su una mutazione del gene MAPT, chiamata p.R406W, che provoca una forma ereditaria di demenza frontotemporale e può simulare una perdita di memoria simile all’Alzheimer. Usando la tecnologia delle cellule staminali, hanno riprogrammato cellule cutanee di pazienti in cellule staminali pluripotenti indotte e poi in grandi quantità di neuroni umani che portavano la mutazione o che erano stati riportati alla normalità tramite editing genetico. Nei neuroni mutanti, Tau totale e Tau fosforilata erano notevolmente aumentati, non perché le cellule producessero più Tau, ma perché la degradavano meno efficacemente. L’imaging super‑risoluzione ha rivelato che quasi tutti i lisosomi nelle cellule mutanti erano pieni di Tau e in particolare presentavano Tau fosforilata che rivestiva la membrana del lisosoma. Questo accumulo indicava che la principale via di smaltimento delle proteine della cellula era bloccata.
Centri di riciclaggio intasati e traffico rallentato
Esaminando più a fondo la macchina del riciclaggio, i ricercatori hanno osservato che i lisosomi nei neuroni mutanti erano più numerosi, più grandi e tendevano a trovarsi più lontano dal corpo cellulare. L’imaging in vivo con coloranti fluorescenti ha mostrato che questi lisosomi si muovevano più lentamente e percorrevano distanze più brevi lungo le fibre nervose, nonostante le piste di microtubuli sottostanti apparissero normali. I neuroni mutanti contenevano anche più autofagosomi, più della proteina adattatrice del carico p62, e goccioline lipidiche extra — segnali che il materiale veniva etichettato per lo smaltimento ma non veniva completamente degradato. Usando un reporter sensibile al pH, hanno trovato che gli autofagosomi nelle cellule mutanti spesso non si fondevano correttamente con i lisosomi, portando a un accumulo di vescicole di riciclaggio «mezza finite» e difetti diffusi nella pulizia cellulare, non solo per Tau ma anche per altri carichi.
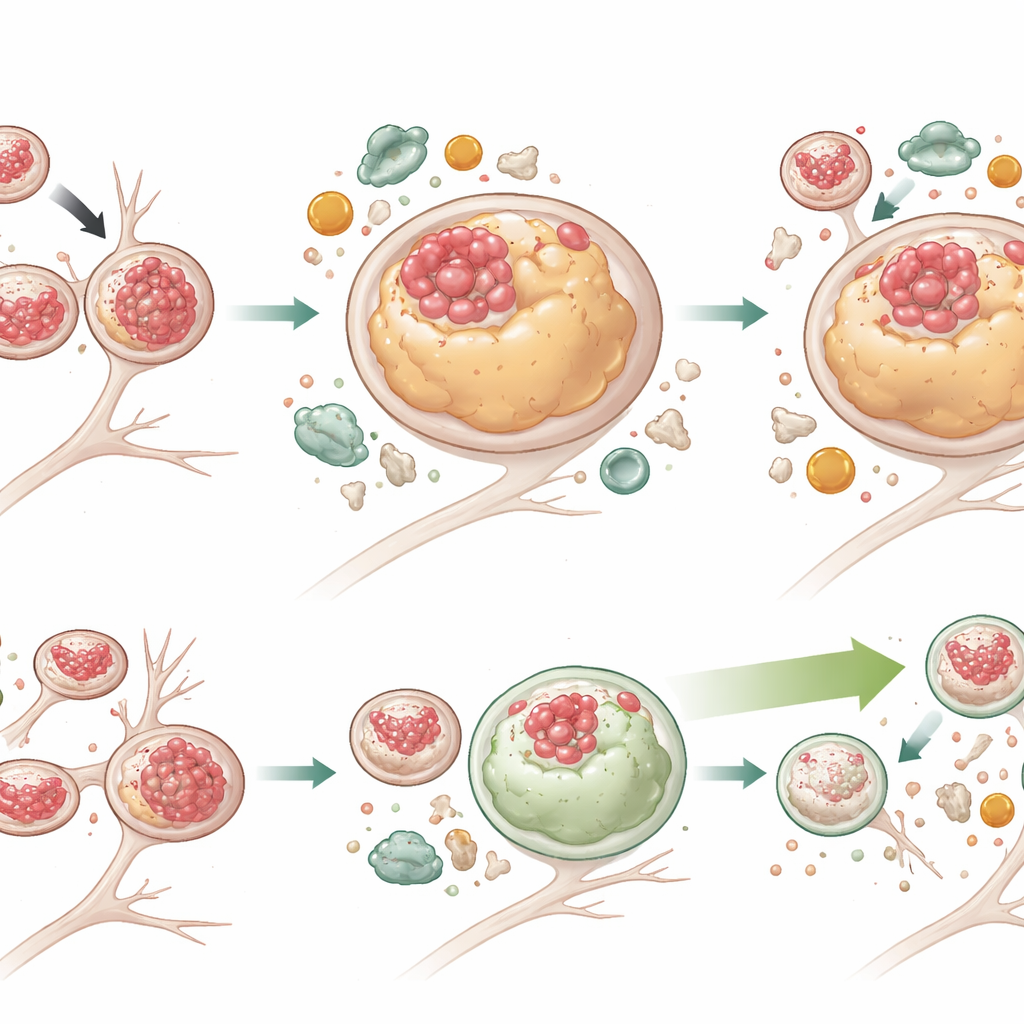
Potenziare la pulizia cellulare senza risolvere l’ingorgo
Per verificare se potenziare l’autofagia potesse superare questi problemi, il team ha trattato i neuroni con G2‑567, una piccola molecola precedentemente dimostrata capace di stimolare il sistema autofagia‑lisosoma. Dopo due settimane di trattamento, i neuroni mutanti presentavano livelli sostanzialmente più bassi sia di Tau totale sia di Tau fosforilata, e molti più lisosomi erano di nuovo privi di Tau. I lisosomi inoltre si ridimensionarono verso le dimensioni normali. I marker di autofagia attiva aumentarono, mentre p62 — un indicatore di degradazione bloccata — diminuì nelle cellule mutanti, dimostrando una più efficace demolizione del carico. Interessante, G2‑567 non correggeva tutti i difetti: i lisosomi nei neuroni mutanti tendevano ancora a trovarsi più lontano dal corpo cellulare e a muoversi lentamente, e una proteina adattatrice (JIP3) legata al trasporto dei lisosomi rimaneva elevata. Ciò suggerisce che il movimento e la funzione degradativa dei lisosomi possono essere parzialmente disaccoppiati, e che migliorare la degradazione da sola può essere sufficiente a ridurre l’accumulo tossico di Tau.
Cosa significa questo per i trattamenti futuri della demenza
Per un pubblico non specialista, la conclusione chiave è che, in questo modello genetico di demenza frontotemporale, il problema non è semplicemente che Tau diventi anormale; è che il sistema di riciclaggio del neurone non riesce a tenere il passo. La mutazione p.R406W di Tau interrompe direttamente diversi passaggi del percorso autofagia‑lisosoma, causando l’accumulo di Tau — specialmente della sua forma fosforilata — sulla e all’interno dei lisosomi, insieme ad altro materiale non degradato. Stimolando farmacologicamente la macchina di pulizia cellulare, i ricercatori sono riusciti a ridurre i livelli di Tau e a normalizzare le dimensioni dei lisosomi, anche se i difetti di trasporto persistevano. Questi risultati rafforzano l’idea che farmaci progettati per potenziare in modo sicuro l’autofagia e la funzione lisosomiale potrebbero aiutare a ripristinare l’equilibrio proteico nelle demenze correlate a Tau e forse in condizioni più comuni come l’Alzheimer.
Citazione: Mirfakhar, F.S., Marsh, J.A., Sato, C. et al. A pathogenic Tau mutation drives autophagy-lysosome dysfunction that limits Tau degradation in a model of frontotemporal dementia. Nat Commun 17, 2699 (2026). https://doi.org/10.1038/s41467-026-70473-5
Parole chiave: proteina tau, autofagia, disfunzione lisosomiale, demenza frontotemporale, neurodegenerazione