Clear Sky Science · it
DiNovo consente il sequenziamento de novo di peptidi ad alta copertura e alta affidabilità tramite proteasi speculari e deep learning
Vedere le proteine con nuovi dettagli
Le proteine sono le piccole macchine che mantengono vive le nostre cellule, ma leggere completamente i loro mattoni costitutivi è ancora sorprendentemente difficile. Questo articolo presenta DiNovo, un nuovo sistema software che aiuta gli scienziati a “leggere” i frammenti proteici in modo molto più completo e affidabile rispetto al passato. Combinando un ingegnoso trucco biochimico con l’intelligenza artificiale moderna, promette di scoprire proteine nascoste, biomarcatori di malattia e persino bersagli immunitari che i metodi tradizionali spesso non rilevano.
Perché leggere i frammenti di proteine è così difficile
La maggior parte delle analisi proteiche moderne si basa sul taglio delle proteine in pezzi più piccoli, detti peptidi, e poi sulla pesatura dei loro frammenti in uno spettrometro di massa. Da questi pesi, i computer tentano di ricostruire la sequenza peptidica originale, come risolvere un cruciverba partendo da indizi parziali. I metodi esistenti di solito assumono che i peptidi provengano da banche dati proteiche note, il che funziona bene per proteine familiari ma fatica con quelle nuove o inattese. Il cosiddetto sequenziamento de novo evita questa limitazione cercando di leggere i peptidi direttamente dai dati, ma spesso non riesce perché alcuni frammenti mancano e alcuni peptidi non vengono mai tagliati in modo netto.
Usare enzimi speculari per colmare i vuoti
L’idea chiave dietro DiNovo è usare coppie di “proteasi speculari” – coppie di enzimi di taglio che incidono le proteine su lati opposti dello stesso tipo di amminoacido. Per esempio, un enzima taglia appena prima di una lisina, mentre il suo partner taglia subito dopo quella lisina. Questo produce due peptidi correlati che condividono lo stesso segmento interno ma hanno estremità differenti. Quando questi peptidi “speculari” vengono analizzati, i loro spettri di massa contengono pattern di frammenti complementari: ciò che manca in uno spettro spesso appare nell’altro. Gli autori mostrano che combinare tali coppie speculari può portare la copertura dei frammenti vicino alla completezza, con circa il 98% dei possibili tagli supportati da segnali sperimentali reali, molto più alto rispetto all’uso di un singolo enzima.
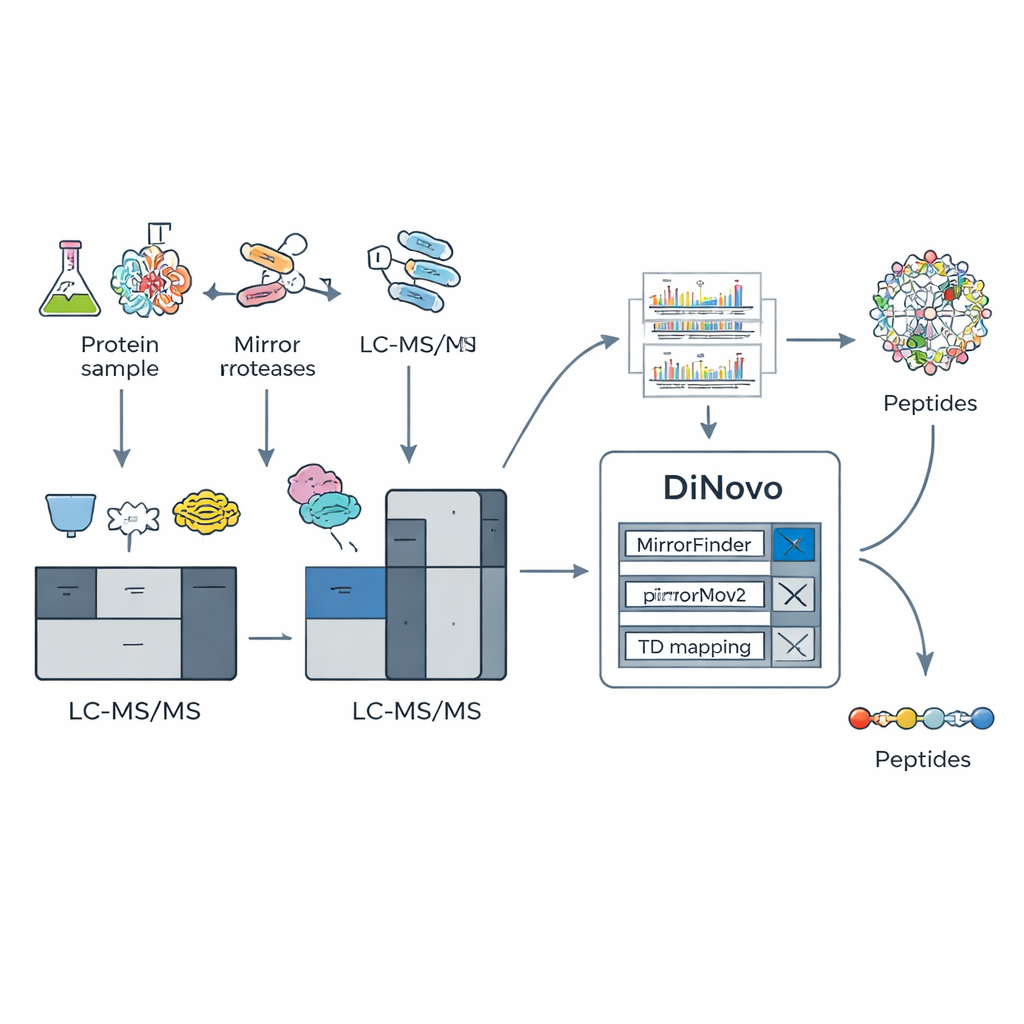
Una pipeline software intelligente progettata per dati speculari
Per sfruttare questo trucco biochimico, il team ha sviluppato DiNovo come workflow software end-to-end. Prima, le proteine provenienti da batteri e lievito vengono digerite con due coppie speculari di enzimi, e i peptidi risultanti sono analizzati mediante spettrometria di massa ad alta risoluzione. DiNovo usa quindi un modulo chiamato MirrorFinder per riconoscere automaticamente quali coppie di spettri derivano da peptidi speculari, facendo questo direttamente dai pattern di segnale piuttosto che da ipotesi di sequenza pregresse. Successivamente, il suo motore de novo principale, MirrorNovo, impiega il deep learning per interpretare quegli spettri accoppiati, mentre un motore di riserva basato su grafi, pNovoM2, offre un’opzione più veloce funzionante su CPU. Insieme, questi strumenti traducono i picchi in sequenze di amminoacidi ed esaminano anche gli spettri individuali che non hanno formato coppie evidenti, estraendo quanta più informazione possibile.
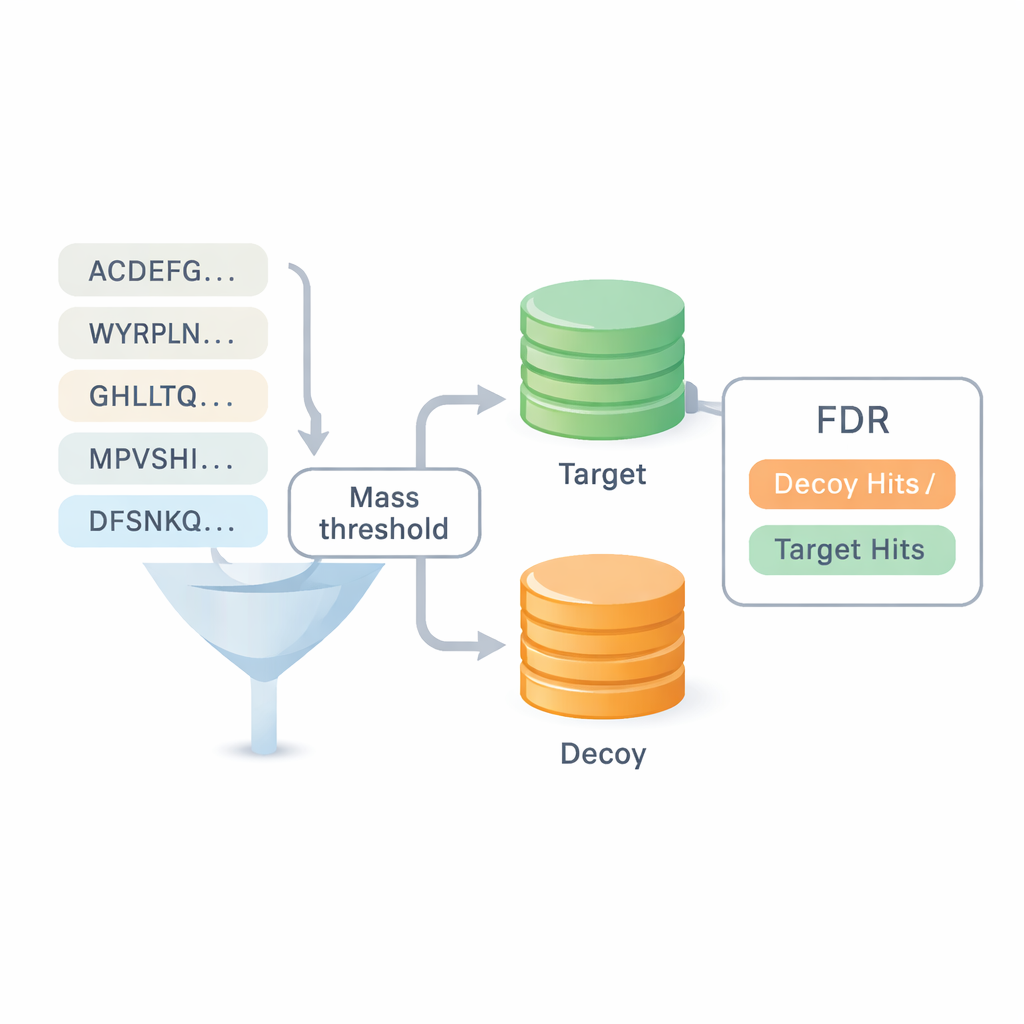
Valutare l’affidabilità senza dipendere dalle vecchie banche dati
Una delle maggiori questioni nel sequenziamento de novo è quanto fidarsi dei risultati. La maggior parte dei benchmark esistenti ricicla risposte da ricerche in banche dati, il che sfuma la linea tra i due approcci e può nascondere errori. DiNovo introduce un metodo di controllo qualità diverso chiamato mappatura target-decoy. Qui, i peptide appena letti vengono mappati su una raccolta combinata di sequenze proteiche reali (target) e artificiose, mescolate (decoy). Confrontando quanto spesso i peptidi vengono assegnati al set reale rispetto a quello mescolato, il software può stimare un tasso di errore, o false discovery rate, senza appoggiarsi a identificazioni precedenti. Questo rende possibile confrontare DiNovo direttamente con i programmi di ricerca in banca dati standard sotto gli stessi criteri di controllo degli errori.
Cosa fornisce DiNovo nella pratica
Nei test su campioni batterici, di lievito e di anticorpi, DiNovo ha letto costantemente molti più peptidi e amminoacidi rispetto ai noti strumenti de novo che utilizzano un solo enzima. Usando due coppie speculari, ha prodotto 2–3 volte più amminoacidi ad alta confidenza rispetto a una configurazione classica con solo tripsina e ha identificato più proteine a livelli di errore simili. In un confronto diretto con tre principali motori di ricerca in banca dati, DiNovo ha trovato numeri analoghi di amminoacidi e proteine, e la maggior parte delle sue sequenze concordava con quelle trovate dai motori di ricerca sugli stessi spettri. Gli autori sostengono che questo livello di copertura e concordanza significa che il sequenziamento de novo, a lungo trattato come metodo di riserva, può oggi stare accanto alla ricerca in banca dati come opzione seria e, in alcuni casi, superiore.
Visione d’insieme: verso una lettura delle proteine completa e non faziosa
Per chi non è specialista, il messaggio è che DiNovo rende molto più semplice leggere con precisione i pezzi proteici senza essere limitati a ciò che è già presente nelle banche dati di riferimento. Raddoppiando o triplicando la quantità di informazioni di sequenza ben supportate e fornendo controlli di errore integrati, questo approccio apre la strada alla scoperta di proteine sconosciute, al tracciamento di variazioni sottili e all’esplorazione di miscele complesse dove molti componenti sono ancora ignoti. In breve, combinando proteasi speculari con deep learning e statistiche rigorose, DiNovo aiuta a trasformare tracce spettrali rumorose in un quadro più chiaro e affidabile delle proteine che sono alla base della salute e della malattia.
Citazione: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Parole chiave: proteomica, sequenziamento de novo di peptidi, spettrometria di massa, deep learning, proteasi speculari