Clear Sky Science · it
Dissezione multimodale della patologia specifica per tipo cellulare di TDP-43 nella corteccia motoria
Perché questa ricerca è importante per le persone
La sclerosi laterale amiotrofica (SLA) e la demenza frontotemporale (FTD) sono malattie cerebrali devastanti che privano le persone del movimento, della parola e della personalità. La maggior parte dei pazienti con SLA e molti con FTD condividono un segno microscopico comune: aggregati di una proteina chiamata TDP-43 che si accumulano dove non dovrebbero. Questo studio pone due domande pratiche con grandi implicazioni per i trattamenti futuri: quali cellule cerebrali sono colpite più gravemente dai problemi di TDP-43 e cosa va storto all'interno di queste cellule a livello di regolazione del DNA e attività genica?
Seguire il danno nel centro del movimento del cervello
I ricercatori si sono concentrati sulla corteccia motoria primaria, la striscia di tessuto cerebrale che controlla il movimento volontario. Utilizzando campioni cerebrali post-mortem donati da persone con SLA, SLA-FTD e controlli neurologicamente sani, hanno isolato nuclei cellulari individuali e hanno letto sia quali geni erano attivi sia quanto fosse compatto il DNA locale. Questo approccio “multi-omico”, applicato a più di 180.000 nuclei, ha permesso di classificare le cellule in tipi precisi: diverse classi di neuroni eccitatori e inibitori, oltre a cellule di supporto come astrociti, oligodendrociti e microglia. Hanno poi combinato questi dati con mappe spaziali dell’espressione genica provenienti da un altro dataset umano per collocare questi tipi cellulari nella nota struttura a strati della corteccia.
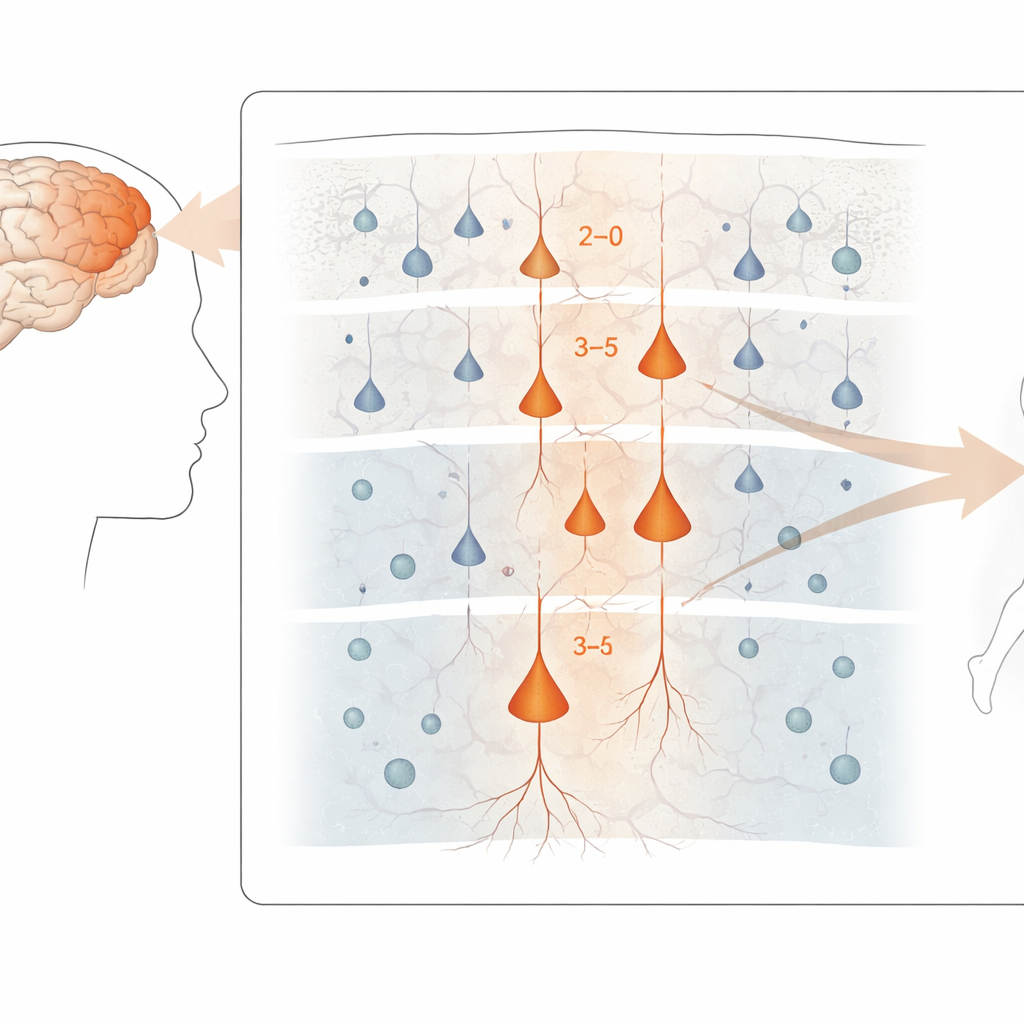
Individuare i neuroni più vulnerabili
In tutta la corteccia motoria, i cambiamenti genici più marcati correlati alla malattia sono emersi nei neuroni eccitatori, le cellule che promuovono l’attività lungo i circuiti cerebrali. In particolare, i neuroni degli strati superficiali e intermedi che connettono all'interno della corteccia, così come alcuni neuroni degli strati profondi che inviano segnali fuori dalla corteccia — incluse le grandi cellule “di Betz” che controllano i motoneuroni spinali — hanno mostrato le alterazioni più pronunciate. Al contrario, gli interneuroni inibitori e molte cellule gliali sono state meno interessate a livello di espressione genica, sebbene alcune mostrassero cambiamenti più sottili. Nonostante questo tumulto molecolare, la composizione complessiva dei principali tipi cellulari nel tessuto era sorprendentemente simile tra pazienti e controlli, suggerendo che il danno riguarda più il modo in cui le cellule funzionano che il semplice numero di cellule andate perdute.
Come TDP-43 rimodella l’attività genica dall’interno
Per separare gli effetti direttamente indotti da TDP-43 da altri processi patologici, il team ha usato una strategia di separazione intelligente. Hanno marcato i nuclei con anticorpi contro TDP-43 e un marcatore neuronale, quindi hanno usato la citometria a flusso per separare i neuroni i cui nuclei avevano perso TDP-43 (un segno di patologia) da quelli che lo avevano conservato. Il sequenziamento di oltre 12.000 di questi nuclei ha rivelato che la perdita di TDP-43 avviene in modo schiacciante nei neuroni eccitatori, in particolare in specifici sottotipi degli strati 2–3, 3–5, 5 e 6. In quei neuroni vulnerabili centinaia di geni risultavano regolati in modo anomalo, inclusi molti già collegati alla SLA. Classiche firme molecolari del malfunzionamento di TDP-43 — come l’apparizione di porzioni “criptiche” nei trascritti genici di STMN2 e KALRN e spostamenti nei siti di processamento degli RNA alle loro estremità — erano chiaramente arricchite nei nuclei carenti di TDP-43.
Rimodellamento epigenetico: non tutto il cambiamento dipende da TDP-43
Poiché hanno misurato sia l’attività genica sia l’accessibilità della cromatina negli stessi nuclei, gli autori hanno potuto chiedere quali cambiamenti erano associati a variazioni nell’impacchettamento del DNA. Hanno trovato decine di migliaia di siti nel genoma in cui l’accessibilità locale della cromatina seguiva l’espressione genica. Molti dei geni alterati in SLA e SLA-FTD si trovavano in queste regioni, indicando che parte della firma di malattia riflette un più ampio rimodellamento epigenetico piuttosto che essere diretta conseguenza della perdita di TDP-43. Interessante, questi cambiamenti legati alla cromatina convergevano spesso su vie di segnalazione coinvolte nella comunicazione cellulare e nella guida degli assoni, ed erano particolarmente pronunciati in certi neuroni eccitatori e negli oligodendrociti. Quando il team ha confrontato i cambiamenti genici associati alla patologia di TDP-43 con quelli legati alle variazioni cromatiniche, ha osservato che si trattava di livelli di alterazione parzialmente sovrapponibili ma per la maggior parte distinti.
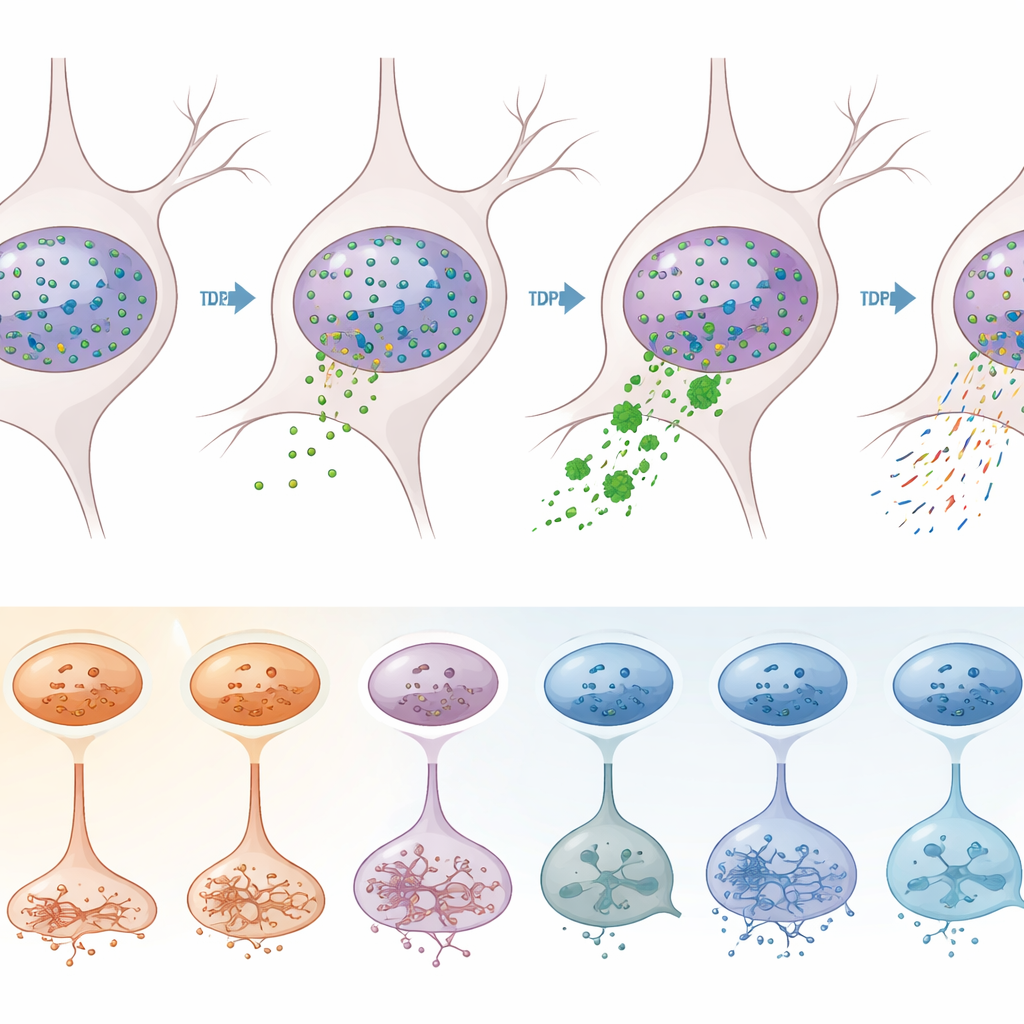
Cosa significa questo per le terapie future
Per un lettore non specialistico, il messaggio chiave è che SLA e SLA-FTD non danneggiano la corteccia motoria in modo uniforme. Colpiscono invece tipi specifici di neuroni eccitatori e, in misura minore, alcune cellule di supporto, alterandone i programmi genici in modi che dipendono sia dal malfunzionamento di TDP-43 sia da più ampie modifiche nel modo in cui il DNA è impacchettato e letto. Questi risultati suggeriscono che trattamenti efficaci potrebbero dover essere specifici per tipo cellulare e per via molecolare — per esempio ripristinando la funzione di TDP-43 o correggendo i suoi errori di splicing nei neuroni più vulnerabili, mentre si interviene separatamente sulle modifiche epigenetiche e sui cambiamenti di segnalazione condivisi da più tipi cellulari. Mappando dettagliatamente questo paesaggio complesso, lo studio fornisce un progetto per progettare interventi più mirati volti a rallentare o prevenire la perdita del controllo del movimento nella SLA e nella SLA-FTD.
Citazione: Ruf, W.P., Kühlwein, J.K., Meier, L. et al. Multi-modal dissection of cell-type specific TDP-43 pathology in the motor cortex. Nat Commun 17, 2406 (2026). https://doi.org/10.1038/s41467-026-69944-6
Parole chiave: SLA, demenza frontotemporale, TDP-43, neuroni della corteccia motoria, multiomica su singolo nucleo