Clear Sky Science · it
Campionamento efficiente di grandi percorsi di transizione e conformazioni intermedie in complessi proteici sub-mesoscopici
Osservare le proteine in movimento
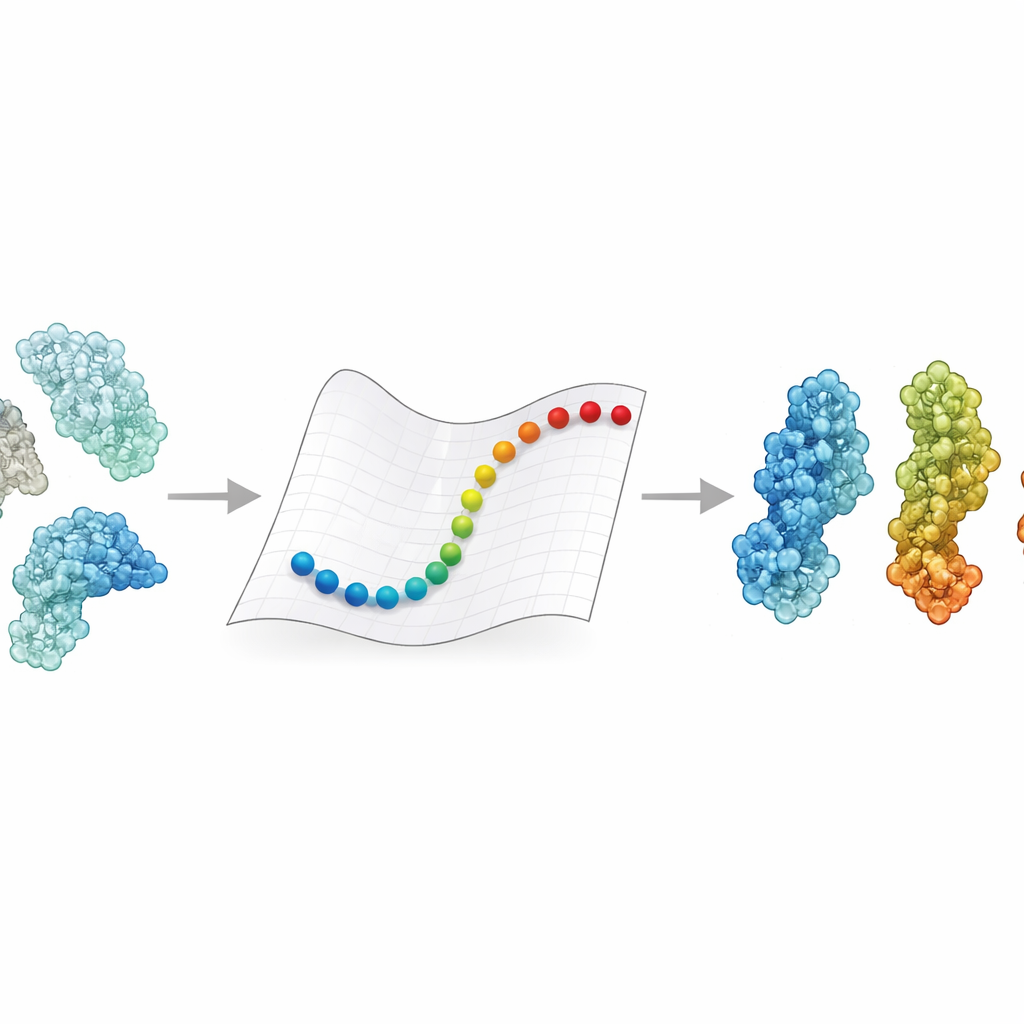
Molte delle molecole che ci mantengono in vita si comportano meno come mattoncini rigidi e più come piccole macchine che cambiano costantemente forma. Questi moti alimentano processi come la produzione di energia, la riparazione del DNA e l’ingresso dei virus nelle cellule. Esperimenti come la crio–microscopia elettronica possono ora congelare alcune di queste conformazioni, ma non i passaggi fugaci intermedi. Questo articolo presenta eBDIMS2, un nuovo metodo computazionale in grado di “riempire i fotogrammi mancanti” del movimento proteico anche per enormi macchine molecolari che in precedenza erano troppo grandi e complesse da simulare su un computer ordinario.
Perché i cambiamenti di forma delle proteine sono importanti
Le proteine raramente restano bloccate in un’unica posa. Si aprono e chiudono, si torcono e si flettono in risposta a segnali come variazioni di potenziale, pH o al legame con una molecola partner. Questi spostamenti possono fare la differenza tra un enzima attivo o inattivo, o tra un recettore che cattura un virus o lo lascia sfuggire. Gli esperimenti ci forniscono istantanee dettagliate di poche conformazioni chiave, e le simulazioni di dinamica molecolare in principio possono collegarle seguendo ogni atomo nel tempo. Ma tracciare tali moti per i grandi complessi ora osservati con la crio–microscopia elettronica, che spesso pesano centinaia di migliaia fino a milioni di Dalton, richiede solitamente supercomputer e settimane di calcolo. Di conseguenza, per molti colossi di interesse medico non sappiamo ancora come uno stato si trasformi in un altro.
Un percorso più veloce attraverso i paesaggi proteici
eBDIMS2 prende una scorciatoia semplificando la rappresentazione delle proteine e il calcolo dei loro moti. Invece di seguire ogni atomo, tratta ogni amminoacido come un singolo punto collegato da molle in una rete elastica. Queste molle catturano come le diverse parti della proteina tendono a muoversi insieme. Il metodo utilizza quindi la dinamica browniana—regole matematiche che imitano il tremolio in un liquido—per spingere la struttura da uno stato sperimentalmente noto verso un altro. Crucialmente, eBDIMS2 presta attenzione solo alle interazioni che contano davvero, usando cutoff di distanza e calcolo parallelo per ridurre i costi. Questo migliora la scalabilità del programma da circa quadratica a quasi lineare rispetto alla dimensione della proteina. In pratica, significa che le transizioni per assemblamenti enormi che si avvicinano a due milioni di Dalton possono essere esplorate in poche ore su un desktop, invece di essere praticamente irraggiungibili.
Confrontare i percorsi con proteine reali
Per verificare se questi percorsi veloci hanno senso biologico, gli autori hanno messo insieme ensemble di 47 grandi proteine e 15 complessi aggiuntivi, per un totale di centinaia di strutture per lo più risolte mediante crio–microscopia elettronica. Hanno usato l’analisi delle componenti principali, uno strumento statistico che individua i modi dominanti in cui ciascuna proteina può muoversi, per organizzare queste strutture in paesaggi conformazionali come aperto, chiuso, attivo o inattivo. eBDIMS2 è stato quindi incaricato di connettere coppie di stati terminali attraverso questo paesaggio. I percorsi risultanti sono stati proiettati sulle stesse mappe a bassa dimensionalità, rivelando se tracciano rotte fluide che passano vicino a intermedi sperimentalmente osservati. In oltre il 30% dei sistemi, le rotte simulate sono passate vicino—a poche angstrom—da strutture intermedie che non erano state fornite come input. Per casi impegnativi come l’enzima di riparazione del DNA DNA-PKcs o la proteina Spike del coronavirus, i percorsi coarse-grained si sovrapponevano inoltre bene a simulazioni a livello atomico molto più costose, incluse dinamiche molecolari mirate e corse avanzate con metodi di campionamento potenziato.
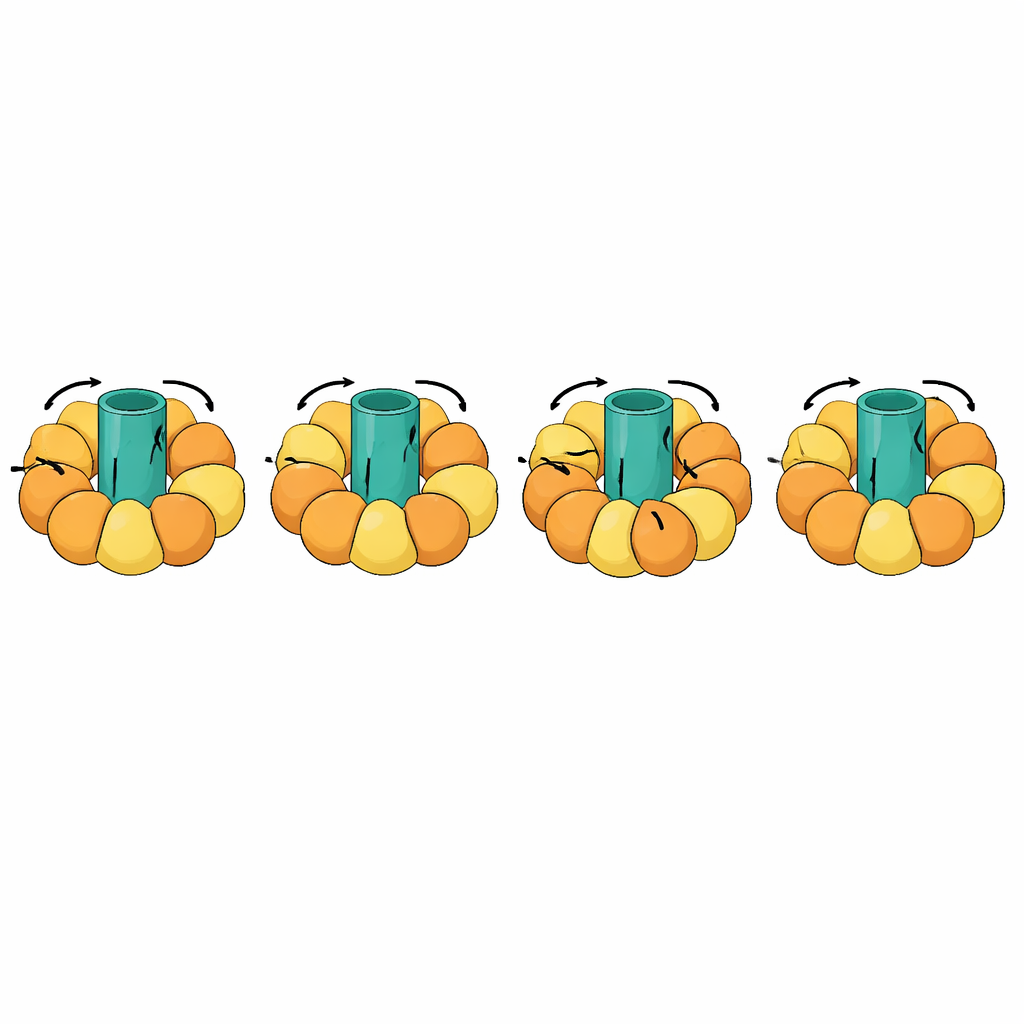
Seguire macchine molecolari giganti
Uno dei test più suggestivi ha coinvolto macchine rotanti come le ATP sintetasi, che producono la moneta energetica della cellula accoppiando un rotore che gira nella membrana a movimenti di apertura e chiusura nelle subunità circostanti. Queste transizioni sono eccezionalmente complesse: parti della molecola devono rimanere rigide e ruotare come un’unità, mentre altre si flettono in un ciclo coreografato. eBDIMS2 introduce un trattamento speciale per questi pezzi quasi rigidi e per modelli sperimentali incompleti con segmenti mancanti, entrambi comuni nella crio–microscopia elettronica. Con queste funzionalità, può simulare cicli rotazionali completi di ATP sintetasi e di altri complessi massivi come chaperoni molecolari, recettori e assemblaggi virali. In tutto il processo, le strutture intermedie generate evitano le gravi distorsioni prodotte da alcuni metodi concorrenti e possono essere convertite in modelli atomistici adatti per calcoli di progettazione di farmaci o per simulazioni più lunghe e dettagliate.
Cosa significa per la biologia e la medicina
Lo studio mostra che eBDIMS2 è in grado di delineare in modo affidabile le principali rotte tra forme proteiche note per sistemi che erano fuori portata per le simulazioni tradizionali. Non sostituisce filmati atomici dettagliati né fornisce energie e tempi precisi, ma offre un modo rapido e fisicamente fondato per mappare come grandi macchine molecolari potrebbero muoversi, usando soltanto una coppia di strutture sperimentali come input. Man mano che i database strutturali si riempiono di molteplici stati di grandi assemblaggi proteici collegati a cancro, infezioni e altre malattie, questo approccio fornisce agli studiosi uno strumento accessibile per collegare i punti, suggerire stati intermedi plausibili e guidare dove indirizzare in seguito metodi ad alta risoluzione o progettazione di farmaci mirata.
Citazione: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Parole chiave: dinamica delle proteine, simulazioni molecolari, cryo-EM, percorsi conformazionali, modellizzazione coarse-grained