Clear Sky Science · it
Attivazione di IRF3 nei cardiomiociti compromette la funzione ossidativa mitocondriale tramite inibizione di PGC-1α e promuove l’insufficienza cardiaca
Perché i cuori stressati e le cellule affaticate contano
L’insufficienza cardiaca è spesso descritta come il cuore che “si consuma”, ma sotto la superficie è anche una storia di infiammazione cronica e di centrali energetiche esauste all’interno delle cellule del muscolo cardiaco. Questo studio pone una domanda apparentemente semplice ma dalle grandi implicazioni: esiste un singolo interruttore molecolare nelle cellule cardiache che collega l’infiammazione dannosa e il cedimento nella produzione di energia — e in tal caso, intervenire su quell’interruttore può cambiare il corso dell’insufficienza cardiaca? Seguendo questo filo, gli autori individuano un attore chiave e mostrano che potenziare moderatamente il programma energetico del cuore può in parte recuperare cuori in insufficienza nei topi.
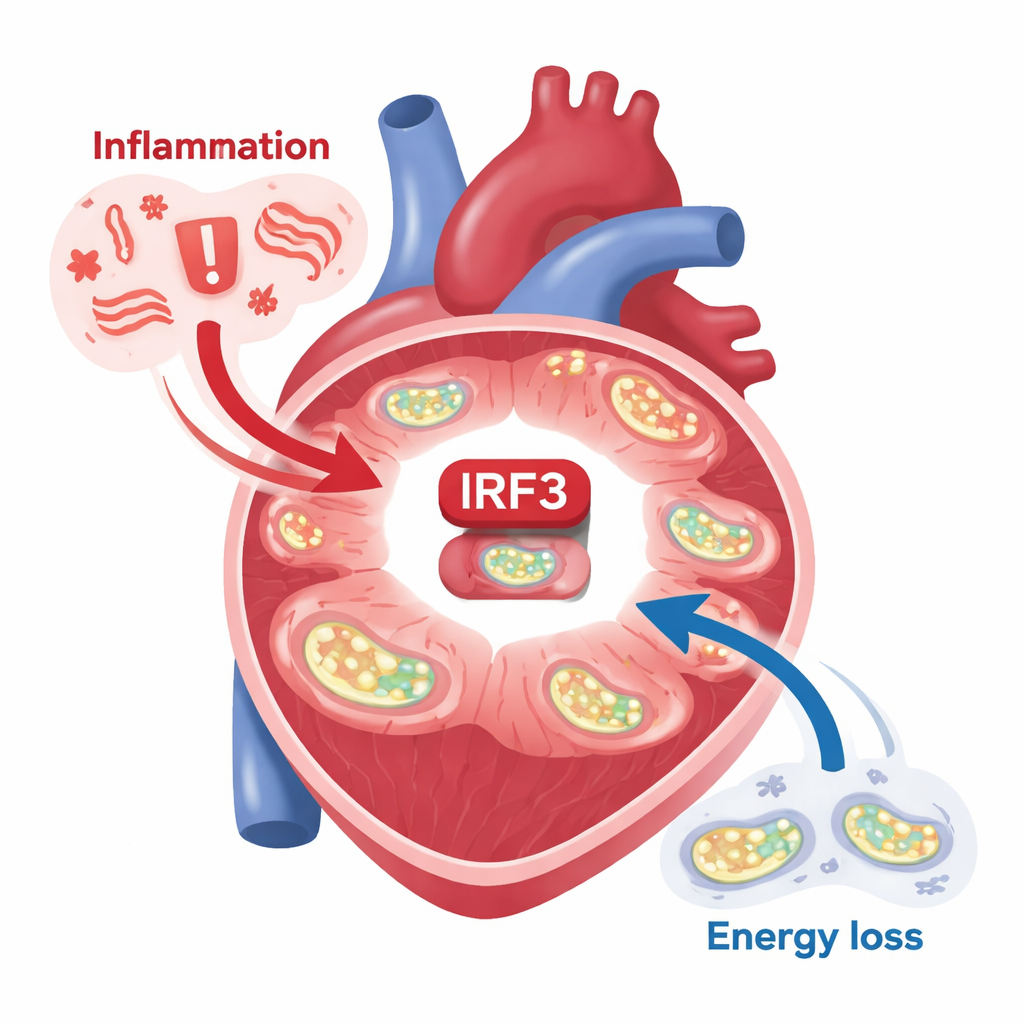
Un interruttore molecolare nei cuori malati
I ricercatori si sono concentrati su una proteina chiamata IRF3, nota soprattutto per aiutare le cellule a rispondere alle infezioni virali. Hanno esaminato tessuto di persone con cardiomiopatia ischemica, una forma comune di insufficienza cardiaca causata da flusso sanguigno ridotto dopo infarti. In questi cuori in scompenso, IRF3 non era solo presente: risultava chimicamente attivata in siti specifici, segno che stava attivamente guidando programmi genici. Allo stesso tempo, i meccanismi che permettono ai mitocondri di trasformare il combustibile in energia tramite la fosforilazione ossidativa erano visibilmente indeboliti. Un pattern simile è emerso nei modelli murini di infarto: quando un’arteria coronaria veniva legata, IRF3 nei cardiomiociti si attivava fortemente e i geni controllati da IRF3 si accendevano. Anche frammenti di DNA mitocondriale — rilasciati da mitocondri danneggiati e che agiscono come segnali di “pericolo” interni — erano sufficienti ad attivare IRF3 in cellule cardiache isolate.
Spegnere IRF3 protegge il cuore
Per verificare se l’attività di IRF3 nei cardiomiociti peggiorasse effettivamente la malattia, il gruppo ha ingegnerizzato topi in cui IRF3 poteva essere rimosso solo dai cardiomiociti, lasciando intatte le altre cellule immunitarie e di supporto. Dopo aver indotto un infarto, questi topi mostravano una funzione contrattile migliore e meno cicatrici rispetto ai topi normali, nonostante lo stesso danno iniziale. In cellule cardiache coltivate in vitro, il silenziamento di IRF3 riduceva l’espressione di geni infiammatori senza alterare altre proteine correlate. Nel loro insieme, questi risultati suggeriscono che IRF3 all’interno della cellula cardiaca non è solo un semplice spettatore: amplifica l’infiammazione e il danno strutturale dopo ischemia e contribuisce alla transizione verso l’insufficienza cardiaca.
Quando IRF3 rimane “acceso”, il sistema energetico crolla
Gli autori hanno quindi invertito l’esperimento: hanno creato topi in cui IRF3 nei cardiomiociti poteva essere forzata in uno stato permanentemente attivo mediante un ingegnoso trucco genetico “fosfomimetico”. Anche senza uno stimolo esterno, questi topi sviluppavano rapidamente grave disfunzione cardiaca, alti livelli di messaggeri infiammatori nel sangue e segni di danno cellulare. Un’analisi approfondita del tessuto cardiaco ha mostrato che, quando IRF3 è attivo cronicamente, sopprime un coordinatore energetico maestro chiamato PGC-1α. Questa molecola normalmente promuove mitocondri sani, l’ossidazione efficiente dei lipidi e un bilancio energetico cellulare equilibrato. Con PGC-1α ridotto, numerose proteine mitocondriali diminuivano, la catena di trasporto degli elettroni vacillava e le scelte metaboliche del cuore cambiavano: scendevano i composti legati all’ossidazione dei grassi come la carnitina, l’uso di chetoni risultava compromesso e il metabolismo del glucosio si alterava. Anche il rapporto NAD⁺/NADH — un indicatore chiave dello stato redox cellulare — si spostava nella direzione sbagliata.
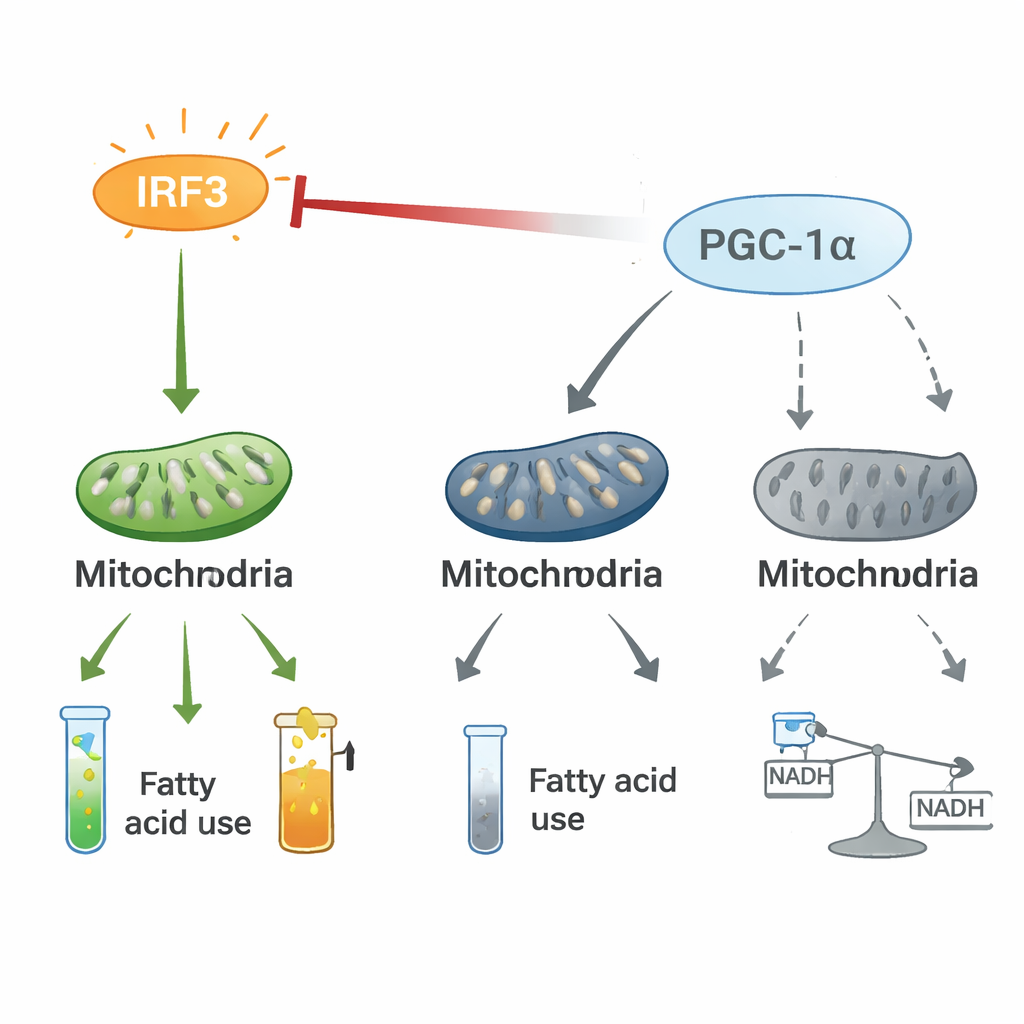
Tira e molla tra infiammazione e controllo energetico
Esperimenti meccanicistici hanno rivelato che IRF3 e PGC-1α costituiscono un asse regolatorio bidirezionale. Nei cardiomiociti, IRF3 attivato si associa fisicamente a PGC-1α e ne attenua la capacità di attivare i geni per l’ossidazione degli acidi grassi. L’abbassamento di IRF3 aumenta i livelli e l’attività di PGC-1α, mentre potenziare PGC-1α smorza i geni infiammatori indotti da IRF3 e ripristina marcatori mitocondriali, anche in condizioni di stress come ipossia o presenza di tossine batteriche. Il tracciamento con isotopi stabili ha mostrato che l’attivazione di IRF3 devia il carbonio dalla normale produzione energetica tramite il ciclo dell’acido citrico verso una via alternativa, la via delle pentoso-fosfato, e interrompe il flusso regolare dei metaboliti. Questo tira e molla tra un interruttore pro-infiammatorio (IRF3) e un copilota energetico (PGC-1α) sembra rimodellare il metabolismo cardiaco in modi che favoriscono infiammazione e perdita di energia.
Ricaricare con cautela le “batterie” del cuore
Infine, il team si è chiesto se aumentare moderatamente PGC-1α potesse controbilanciare il danno indotto da IRF3. Hanno usato un vettore di terapia genica mirato al cuore per innalzare PGC-1α in modo moderato — ma non eccessivo — negli stessi topi con IRF3 iperattivo. Questo lieve aumento ha migliorato la funzione di pompaggio, incrementato le proteine mitocondriali, potenziato i geni per l’ossidazione dei grassi e il metabolismo del NAD, e ridotto l’attività genica infiammatoria e fibrotica. Negli esperimenti cellulari, l’espressione congiunta di PGC-1α con IRF3 attivo ha ripristinato un rapporto NAD⁺/NADH più sano e ha riportato l’uso dei carburanti verso i lipidi. Per il lettore non specialista, ciò significa che ricaricare con cura il “sistema di gestione della batteria” del cuore può compensare in parte gli effetti nocivi di un interruttore infiammatorio cellulare cronicamente acceso.
Cosa significa per la cura futura dell’insufficienza cardiaca
Questo lavoro pone IRF3 come un collegamento centrale tra infiammazione e collasso energetico all’interno dei cardiomiociti. Invece di considerare infiammazione e metabolismo come problemi separati nell’insufficienza cardiaca, lo studio suggerisce che sono intrecciati tramite un asse IRF3–PGC-1α. Sebbene questi risultati provengano da topi e da modelli cellulari, aprono la possibilità che terapie future possano sia ridurre l’attività di IRF3 sia rafforzare PGC-1α e la funzione mitocondriale per rallentare o prevenire l’insufficienza cardiaca dopo un infarto. In termini semplici, calmare un sistema di allarme cellulare iperattivo e sostenere le fabbriche energetiche del cuore potrebbe rivelarsi una strategia combinata potente per mantenere più a lungo i cuori indeboliti in grado di battere con forza.
Citazione: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Parole chiave: insufficienza cardiaca, infiammazione, mitocondri, cardiomiociti, PGC-1α