Clear Sky Science · it
XL-MSDigger: una soluzione versatile basata sul deep learning per la spettrometria di massa con cross-linking
Vedere come le proteine si tengono insieme
Ogni processo nel nostro corpo dipende non solo dal corretto ripiegamento delle proteine, ma anche dal fatto che trovino i partner giusti. Tuttavia osservare queste relazioni molecolari in azione è notoriamente difficile. Questo studio presenta XL-MSDigger, una piattaforma software che utilizza l’intelligenza artificiale moderna per estrarre segnali molto più chiari da una tecnica sperimentale rumorosa chiamata spettrometria di massa con cross-linking, aiutando gli scienziati a mappare come le proteine sono disposte e con chi interagiscono all’interno delle cellule.
Districare un mondo molecolare affollato
Per comprendere come sono costruite le proteine e come si connettono, i ricercatori spesso ricorrono alla spettrometria di massa con cross-linking. In questo approccio, piccoli “ponti” chimici legano tra loro regioni prossime di proteine. I pezzi legati vengono quindi frammentati e pesati in uno spettrometro di massa. In teoria, il pattern dei frammenti rivela quali porzioni di proteina erano vicine nello spazio, come trovare quali pagine di un libro erano appuntate insieme. In pratica, però, i dati risultanti sono estremamente complessi. Gli strumenti informatici esistenti si basano per lo più sulle informazioni di massa di base e faticano con l’enorme numero di possibili combinazioni, portando a connessioni mancate e corrispondenze spurie.
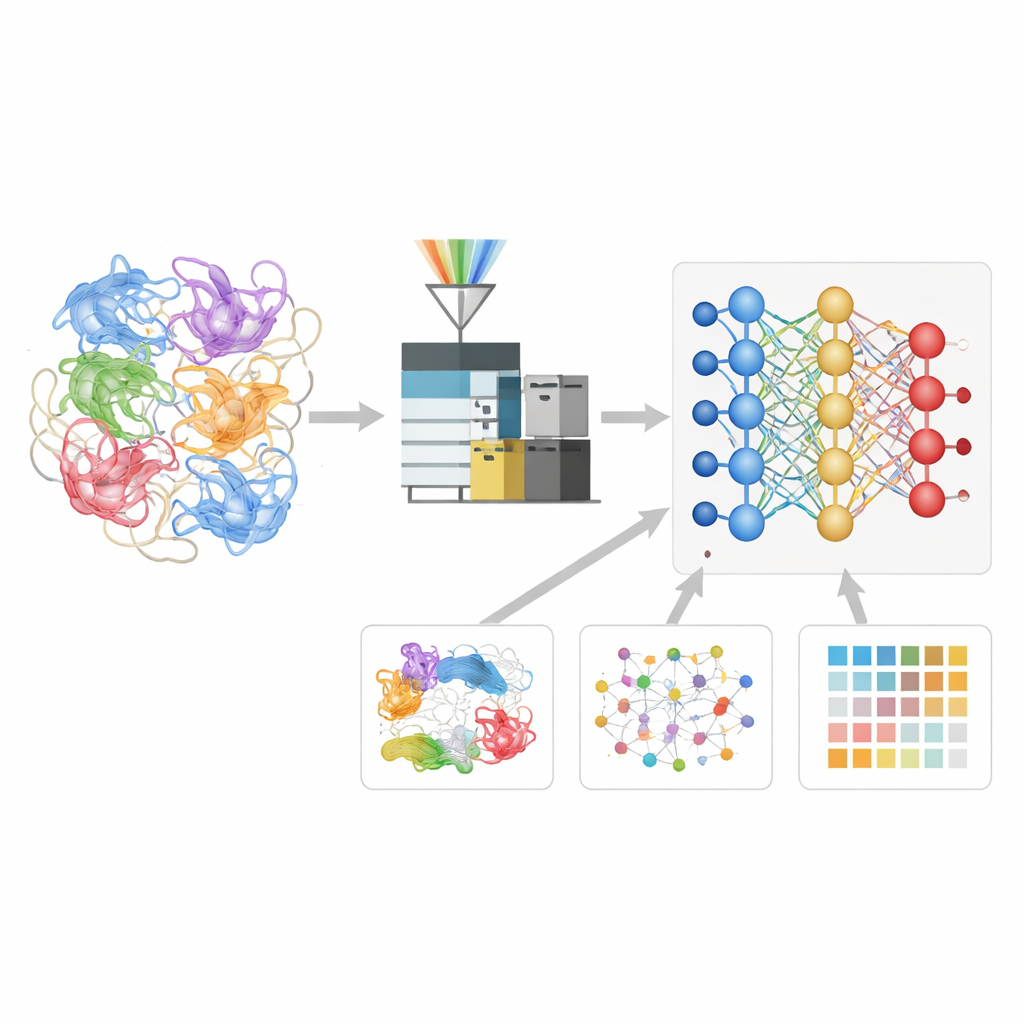
Insegnare a una rete neurale il linguaggio dei frammenti proteici
Gli autori hanno costruito un modello di deep learning chiamato Deep4D-XL per interpretare meglio questi esperimenti di cross-linking. Hanno prima creato un ampio set di riferimento cross-linkando proteine provenienti da cellule umane, frammentandole in peptidi e registrando non solo le loro masse ma anche il tempo di volo nello strumento e il comportamento nella camera di ion-mobility. Ogni coppia cross-linkata è stata codificata per il modello, che usa un design siamese a due bracci per leggere entrambi i peptidi partner e un passaggio di cross-attention per combinare le loro informazioni. Da questo, la rete impara a prevedere tre proprietà chiave di qualsiasi nuovo peptide cross-linkato: quando dovrebbe comparire nell’esperimento, come dovrebbe muoversi e quale dovrebbe essere il suo pattern di frammentazione.
Trasformare le previsioni in segnali più puliti
XL-MSDigger incapsula questo motore di previsione in flussi di analisi per due principali modalità di acquisizione dei dati. Nello stile tradizionale, mirato, lo strumento registra selettivamente i frammenti dagli ioni scelti in tempo reale. XL-MSDigger prende le corrispondenze iniziali dal software di ricerca consolidato e le rivaluta usando il comportamento previsto dal modello per ciascun candidato. Una seconda rete neurale confronta previsione ed esperimento su più dimensioni e assegna punteggi migliorati. Questo passaggio di ricalcolo quasi raddoppia il numero di legami tra proteine diversi rilevati con fiducia in campioni di lievito e umano, mantenendo bassi i tassi di errore e rivelando molte più interazioni proteina–proteina rispetto a prima.
Interpretare ondate di dati non selettivi
Un modo più recente di far funzionare questi strumenti, chiamato acquisizione indipendente dai dati (DIA), registra i frammenti di quasi tutto ciò che è nel campione, migliorando la copertura ma generando dati travolgenti. Fino ad ora non esisteva un buon metodo per stimare quante delle cross-link risultanti fossero veramente reali. XL-MSDigger utilizza Deep4D-XL per costruire una libreria di “decoy” attentamente abbinata di cross-link finti, quindi analizza insieme voci reali e decoy. Osservando quanto spesso i decoy passano i filtri, il software può stimare il tasso di false discovery e addestrare un’altra rete neurale a separare corrispondenze vere da false. Questo ricalcolo aumenta di circa cinque volte il numero di segnali cross-link affidabili e produce una chiara separazione tra pattern reali e decoy.
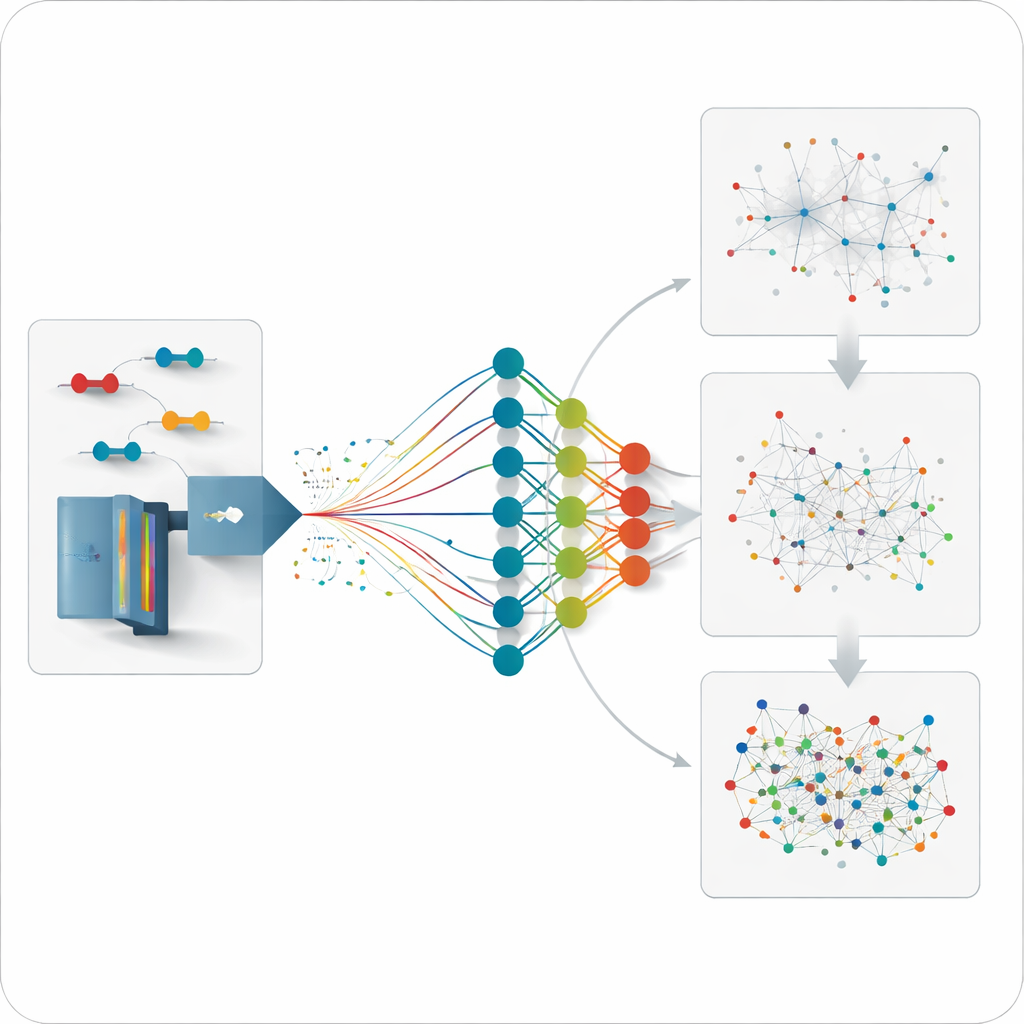
Prevedere ciò che non è stato ancora misurato
Poiché il modello può prevedere come si comporterà qualsiasi plausibile peptide cross-linkato, il team può spingersi oltre e analizzare dati per legami che non sono mai stati misurati direttamente prima. Generano librerie previste di dimensioni moderate focalizzate su proteine selezionate o su reti di interazione e poi cercano i dati non selettivi contro queste librerie. Questa strategia scopre legami aggiuntivi all’interno di singole proteine e tra partner di importanti proteine chaperone, con distanze che concordano bene con strutture tridimensionali note. Recupera inoltre interazioni perse dalle librerie sperimentali tradizionali e più limitate, specialmente per connessioni a bassa abbondanza.
Aprire una finestra più chiara sulle collaborazioni proteiche
Per i non specialisti, il messaggio chiave è che XL-MSDigger agisce come un riconoscitore di pattern altamente addestrato sovrapposto a un metodo sperimentale già potente. Imparando come dovrebbero apparire segnali cross-link genuini su più dimensioni contemporaneamente, può setacciare enormi dataset disordinati, scartare probabili impostori e recuperare connessioni proteiche reali ma precedentemente nascoste. Sebbene le applicazioni su interi proteomi richiederanno ancora elevate risorse di calcolo, questo lavoro dimostra che combinare esperimenti di cross-linking con il deep learning può affinare notevolmente la nostra visione di come le proteine sono disposte e con chi si incontrano all’interno della cellula.
Citazione: Chen, M., Hao, Y., Huang, X. et al. XL-MSDigger: a deep learning-based, versatile solution for cross-linking mass spectrometry. Nat Commun 17, 2554 (2026). https://doi.org/10.1038/s41467-026-69489-8
Parole chiave: interazioni proteiche, spettrometria di massa con cross-linking, deep learning, proteomica, acquisizione indipendente dai dati