Clear Sky Science · it
Un riferimento generico definito da picchi di consenso per l'analisi dei dati single-cell ATAC-seq
Perché mappare le porte aperte del nostro DNA è importante
Ogni cellula del corpo contiene essenzialmente lo stesso DNA, eppure le cellule cerebrali, del sangue e tumorali si comportano in modo molto diverso. Una ragione chiave è che solo alcuni tratti di DNA sono esposti e "aperti" per l'utilizzo in un dato momento. Le nuove tecnologie single-cell possono ora misurare questa apertura a livello genomico, ma finora mancavano di una mappa di riferimento comune—qualcosa come un atlante standard—per confrontare i risultati tra esperimenti e laboratori. Questo studio costruisce una tale mappa, chiamata cPeaks, e mostra come possa affinare la nostra visione dei tipi cellulari, dello sviluppo e del cancro.
Trasformare molti esperimenti in una mappa condivisa
Gli autori hanno iniziato raccogliendo 624 esperimenti di alta qualità che misuravano la cromatina aperta—le parti accessibili del DNA—in più di 40 organi umani. In ciascun esperimento, programmi informatici avevano già segnalato "picchi" dove il DNA era particolarmente esposto. Invece di trattare ogni dataset separatamente, il team ha sovrapposto con cura tutte queste liste di picchi lungo il genoma e ha unito le regioni sovrapposte. Hanno quindi esaminato quanto spesso ogni piccola posizione all'interno di queste regioni unite veniva chiamata aperta negli esperimenti, trasformando ciascuna regione in una forma caratteristica che rifletteva quanto fosse consistente la sua comparsa. Quando una regione unita conteneva in realtà diversi siti aperti ravvicinati, l'hanno divisa in più unità semplici. Queste unità—circa 1,4 milioni in totale—sono divenute i picchi di consenso osservati, o cPeaks, un catalogo di riferimento candidato per l'accessibilità della cromatina umana. 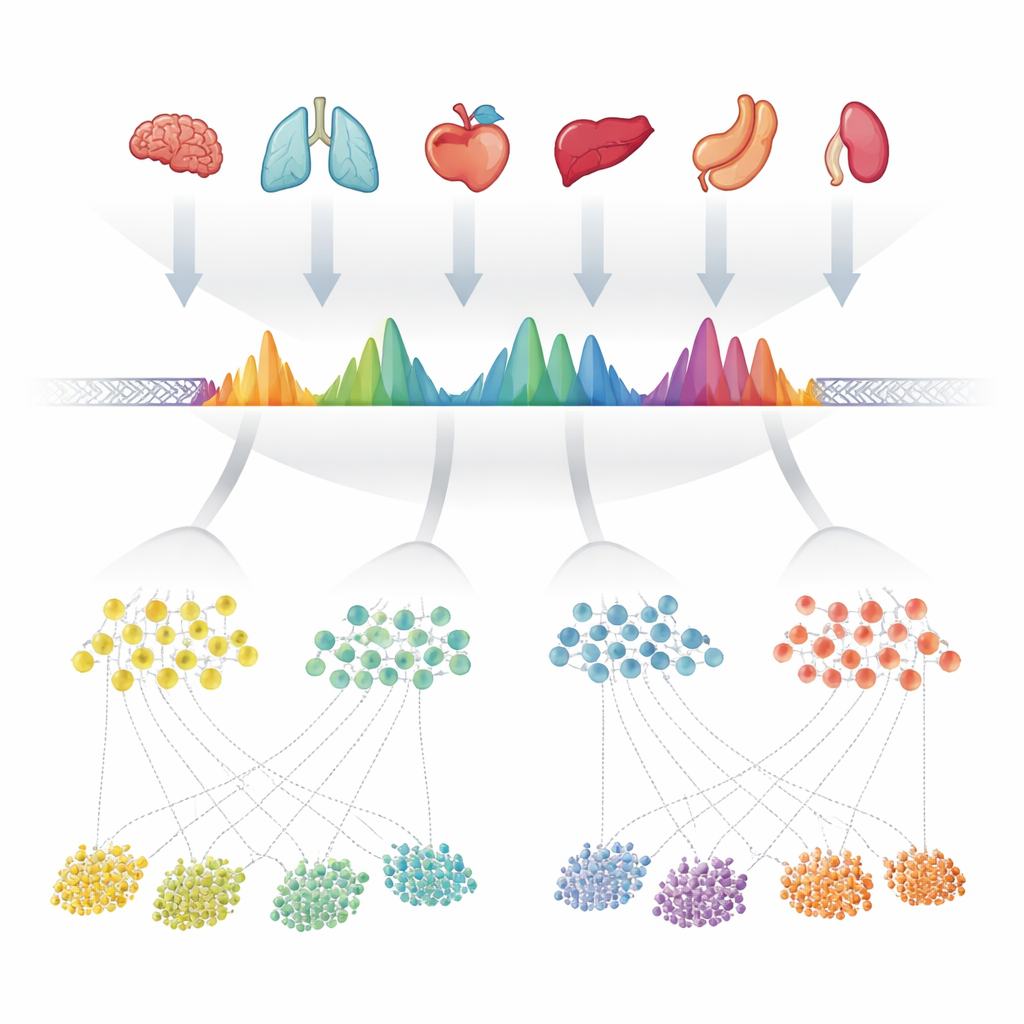
Un'impronta stabile attraverso tessuti e tecnologie
Per essere un riferimento utile, questi cPeaks devono rappresentare caratteristiche genuine e ripetibili del genoma, non stranezze di campioni o software particolari. Gli autori hanno testato questo ricreando le loro regioni unite usando solo campioni di sangue, solo tessuti solidi, banche dati pubbliche separate e persino diversi metodi di laboratorio per sondare il DNA aperto. In ogni caso, le stesse posizioni genomiche hanno prodotto forme di picco notevolmente simili, e la maggior parte dei dataset single-cell esaminati sovrapponeva oltre il 90% dei propri picchi con il catalogo cPeaks. Le letture provenienti da molti organi si accumulavano precisamente attorno ai centri dei cPeaks, dimostrando che queste regioni catturano in modo affidabile dove la cromatina è aperta. Rispetto a insiemi di riferimento precedenti basati su tecnologie correlate, i cPeaks coprivano una porzione maggiore del DNA accessibile rilevato dagli esperimenti ATAC-seq e catturavano quasi tanto segnale quanto i picchi definiti ex novo in ciascun dataset—nonostante fossero fissi e riutilizzabili.
Insegnare a una rete neurale a trovare regioni mancanti
Anche centinaia di campioni esistenti non possono coprire ogni possibile tipo cellulare. Per estendere la loro mappa in regioni non ancora osservate, il team ha fatto ricorso al deep learning. Hanno addestrato una rete neurale convoluzionale unidimensionale sulle sequenze di DNA: esempi che ricadevano all'interno dei cPeaks osservati sono serviti come positivi, mentre regioni di sfondo scelte casualmente come negativi. Il modello ha imparato a distinguere i due con alta accuratezza, implicando che i cPeaks portano modelli di sequenza riconoscibili. Quando i ricercatori hanno nascosto deliberatamente i picchi specifici di un tessuto per volta, la rete li ha comunque recuperati dalla sola sequenza, inclusi siti rari e specifici di tessuto. Hanno poi fatto scorrere una finestra piccola lungo il resto del genoma, valutando ogni segmento e aggiungendo circa 280.000 nuove regioni ad alto punteggio al catalogo come cPeaks predetti, migliorando in particolare la copertura nei tessuti sottorappresentati nei dati originali.
Collegare le regioni aperte a geni, tipi cellulari e cellule rare
Con un riferimento più ricco a disposizione, gli autori hanno indagato cosa fanno queste regioni. Molti cPeaks si trovano vicino ai siti di inizio e fine dei geni o sovrappongono elementi regolatori noti come promotori, enhancer e siti di legame per proteine architetturali come CTCF. Una piccola sottoserie è accessibile in quasi tutti i dataset; questi cPeaks "housekeeping" più estesi tendono a trovarsi in regioni di promotore centrale di geni necessari per la manutenzione cellulare di base. Il team ha anche classificato i cPeaks in base a quanto nitidi e consistenti sono i loro bordi attraverso i campioni, caratteristica che riflette quanto precisamente il DNA vicino è impacchettato in nucleosomi. Le regioni con confini ben definiti sono arricchite per particolari famiglie di fattori di trascrizione noti per rimodellare la cromatina e guidare lo sviluppo. Quando i cPeaks sono stati usati come insieme di feature per analizzare più dataset single-cell, hanno migliorato l'accuratezza dell'annotazione dei tipi cellulari, e sono stati particolarmente utili per identificare tipi cellulari rari e sottotipi sottili che insiemi di picchi precedenti o semplici griglie genomiche spesso confondevano.
Seguire lo sviluppo e il cancro usando un linguaggio comune
La potenza di un riferimento standard diventa evidente confrontando contesti biologici molto diversi. Usando i cPeaks, gli autori hanno rianalizzato dati single-cell della retina umana in sviluppo, grandi atlanti di tessuti fetali e adulti, e diversi tumori. Hanno potuto ricostruire traiettorie di sviluppo e osservare che la frazione di cPeaks con confini netti e "ben posizionati" tende ad aumentare durante le fasi di transizione, per poi diminuire man mano che le cellule si consolidano in identità stabili. Un pattern simile è apparso attraverso gli stadi tumorali: i tumori intermedi mostravano una proporzione maggiore di queste regioni strutturate, suggerendo un intenso rimodellamento regolatorio. In un tumore ovarico, i cPeaks hanno aiutato a rivelare due sottoclonalità cancerose distinte con differenti variazioni del numero di copie del DNA, mostrando come il riferimento possa mettere in luce complessità nascoste nella malattia.
Cosa significa questo per la ricerca genomica futura
Per i non specialisti, i cPeaks possono essere considerati come un insieme standardizzato di coordinate che segnano dove il genoma è più probabilmente fisicamente aperto e attivo in molti tipi cellulari umani. Allineando nuovi esperimenti single-cell sulla cromatina a questa mappa condivisa, i ricercatori possono confrontare i risultati tra studi, individuare più facilmente stati cellulari rari o di transizione e cominciare a costruire modelli su larga scala della regolazione genica—proprio come i cataloghi di geni standardizzati hanno reso possibile l'emergere di atlanti single-cell di RNA. L'attuale catalogo cPeak è una prima bozza che crescerà con l'arrivo di nuovi dati, ma fornisce già un linguaggio comune per descrivere l'accessibilità della cromatina, avvicinandoci a una visione unificata di come il confezionamento del DNA guidi sviluppo, salute e malattia. 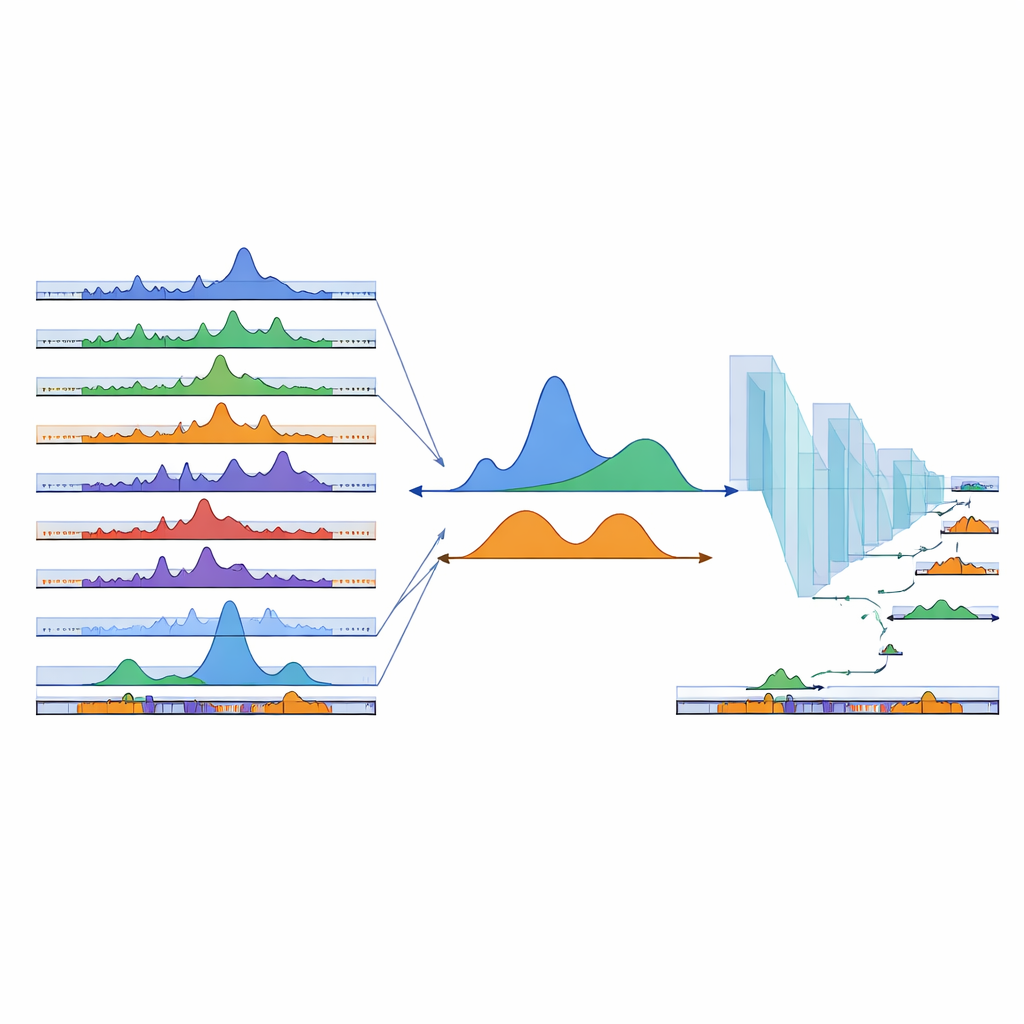
Citazione: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
Parole chiave: accessibilità della cromatina, single-cell ATAC-seq, picchi di consenso, regolazione genica, genomica con deep learning