Clear Sky Science · it
Mirare la NHEJ attiva la segnalazione STING tramite la degradazione di MYC per potenziare l’immunità antitumorale nel SCLC
Perché questa ricerca è importante
Il carcinoma polmonare a piccole cellule è uno dei tumori più letali: la maggior parte dei pazienti vive meno di un anno dalla diagnosi. Paradossalmente, questi tumori presentano numerose mutazioni del DNA che, in teoria, dovrebbero renderli bersagli facili per il sistema immunitario, ma nella pratica rispondono poco alle terapie immunitarie moderne. Questo studio individua un freno molecolare nascosto che impedisce al sistema immunitario di riconoscere questi tumori e mostra come disattivando una proteina chiave della riparazione del DNA sia possibile trasformare questi tumori da “freddi” a “caldi”, consentendo alle terapie esistenti di funzionare molto meglio.
Un interruttore di riparazione nascosto nei tumori polmonari
I ricercatori hanno iniziato esaminando dati genetici provenienti da oltre 179.000 tumori umani in 24 tipi di cancro. Si sono concentrati su una via di riparazione del DNA chiamata non-homologous end joining, che ripara rotture pericolose nelle doppie eliche. Un controllore centrale di questa via, una proteina nota come DNAPKcs (codificata dal gene PRKDC), è risultata insolitamente espressa nel carcinoma polmonare a piccole cellule. Tra migliaia di campioni di tumore polmonare, i casi di piccolo cellule mostravano l’attività più elevata di questo interruttore di riparazione. I pazienti i cui tumori avevano livelli più alti di PRKDC vivevano meno a lungo e traevano meno beneficio dalla chemioterapia standard e dagli inibitori dei checkpoint immunitari, suggerendo che DNAPKcs aiuta i tumori a sopravvivere sia al danno al DNA sia all’attacco immunitario.
Dal danno al DNA a un allarme interno
Per capire cosa succede quando questo interruttore di riparazione viene spento, il gruppo ha usato farmaci e strumenti di silenziamento genico per bloccare DNAPKcs in pannelli di cellule di carcinoma polmonare a piccole cellule e in modelli tumorali murini. In molti di questi modelli, specialmente in quelli che ricordano sottotipi umani con alta attività dell’oncogene MYC, gli inibitori di DNAPKcs hanno ridotto drastica-mente la crescita delle cellule tumorali e hanno persino fatto regredire tumori derivati da pazienti nei topi. A livello cellulare, il blocco di DNAPKcs ha portato a un accumulo di DNA rotto, visibile come puntiforme marker di danno nel nucleo e come piccole strutture extra piene di DNA chiamate micronuclei. Questi frammenti di DNA sono fuoriusciti nel citoplasma, dove possono essere riconosciuti come segnali di pericolo.
Accendere l’allarme “virale” della cellula
Il DNA libero nel posto sbagliato è normalmente un segnale di infezione virale. Le cellule lo rilevano tramite un sensore chiamato cGAS, che attiva una via di allarme a valle chiamata STING. Gli autori hanno mostrato che dopo l’inibizione di DNAPKcs, cGAS si concentra sui micronuclei, STING viene attivato e si accende una cascata di molecole che stimolano l’immunità. Le cellule producono più interferoni di tipo I e II e chemiotassi che attraggono cellule immunitarie. Aumenta anche l’esposizione sulla superficie di proteine “distintive” chiave (molecole MHC di classe I), che aiutano le cellule immunitarie a riconoscere gli antigeni tumorali. Quando la via STING è stata bloccata chimicamente o silenziata geneticamente, questi cambiamenti sono in gran parte scomparsi e gli effetti antitumorali dell’inibizione di DNAPKcs sono risultati molto più deboli, sottolineando che questo sistema di allarme interno è essenziale per la risposta.
Disarmare MYC per smascherare il tumore
Lo studio collega inoltre DNAPKcs al potente driver della crescita MYC, una proteina da lungo tempo considerata “non drogabile”. Nei tumori con alta attività di MYC, l’inibizione di DNAPKcs ha ridotto la segnalazione attiva di AKT e ha rilasciato un freno molecolare su un’altra chinasi, GSK3β. Una volta attivata, GSK3β ha etichettato MYC per la distruzione, determinando una diminuzione dei livelli di proteina MYC. L’abbassamento diretto di MYC mediante strumenti genetici ha imitato molti degli effetti di attivazione immunitaria del blocco di DNAPKcs: la segnalazione STING è aumentata, i geni degli interferoni si sono attivati e l’MHC di classe I è aumentato. Al contrario, costringere le cellule a sovraesprimere MYC ha in gran parte annullato l’impatto immunostimolante dell’inibitore di DNAPKcs. Questi risultati suggeriscono che DNAPKcs normalmente contribuisce a stabilizzare MYC e che indurre la degradazione di MYC è un passaggio chiave per risvegliare l’immunità antitumorale.
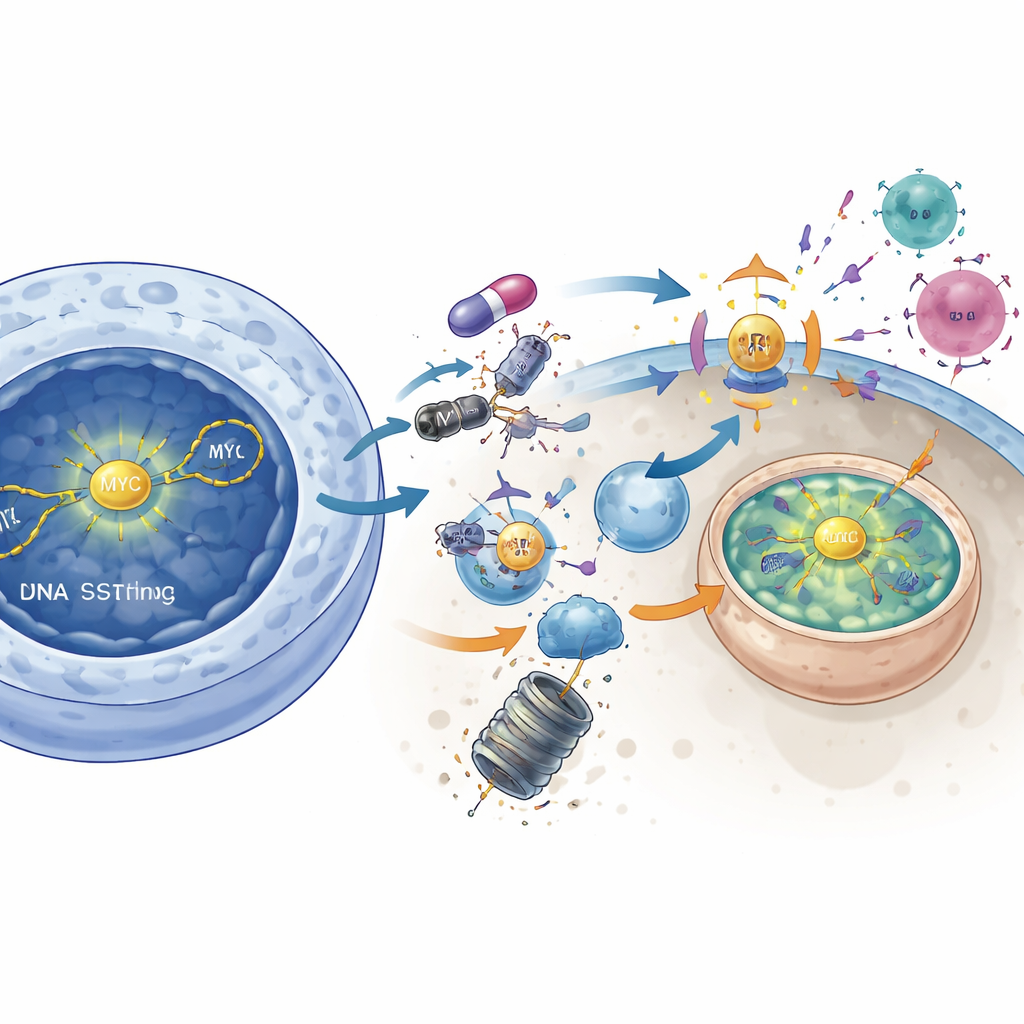
Da tumori “freddi” a “caldi” nei modelli in vivo
In modelli murini immunocompetenti che assomigliano strettamente al carcinoma polmonare a piccole cellule umano, il trattamento con un inibitore di DNAPKcs da solo ha rallentato o ridotto significativamente i tumori. Importante, la combinazione dell’inibitore con un farmaco anti–PD-L1 esistente ha trasformato tumori precedentemente resistenti, portando a regressioni tumorali drammatiche e, in alcuni casi, alla scomparsa completa. Il profilo dettagliato del sistema immunitario ha mostrato che l’inibizione di DNAPKcs aumentava le cellule CD8 T con attività citotossica, potenziava i macrofagi pro-infiammatori M1, riduceva le popolazioni di cellule T soppressive e innalzava i livelli di MHC di classe I nei tumori. La rimozione delle cellule CD8 o la disattivazione di STING hanno invertito questi benefici, confermando che la terapia funziona trasformando il tumore in un faro per l’attacco immunitario più che agire solo con l’uccisione diretta delle cellule tumorali.
Cosa significa per i pazienti
Complessivamente, questi risultati rivelano DNAPKcs come un coordinatore centrale sia della riparazione del DNA sia dell’evasione immunitaria nel carcinoma polmonare a piccole cellule. Bloccando DNAPKcs, i tumori accumulano danno al DNA, MYC si destabilizza, l’allarme cGAS–STING viene attivato e le vie degli interferoni e della presentazione degli antigeni si accendono. Questa catena di eventi converte tumori silenti dal punto di vista immunitario in tumori che rispondono in modo significativo al blocco dei checkpoint e alla chemioterapia nei modelli preclinici. Pur richiedendo ancora studi clinici, il lavoro suggerisce che gli inibitori di DNAPKcs già disponibili potrebbero essere combinati con l’immunoterapia per offrire ai pazienti affetti da questo cancro aggressivo maggiori possibilità di un controllo duraturo.
Citazione: Chakraborty, S., Elliott, A., Sen, U. et al. Targeting NHEJ activates STING signaling through MYC degradation to boost antitumor immunity in SCLC. Nat Commun 17, 2597 (2026). https://doi.org/10.1038/s41467-026-69262-x
Parole chiave: carcinoma polmonare a piccole cellule, inibizione della riparazione del DNA, via STING, degradazione di MYC, immunoterapia tumorale