Clear Sky Science · it
Un trasformatore equivariant preaddestrato per un apprendimento unificato della rappresentazione molecolare 3D
Insegnare ai computer a vedere le molecole in 3D
Progettare nuovi farmaci e materiali dipende dalla comprensione di come le molecole appaiono e si muovono realmente nello spazio tridimensionale, non solo come formule bidimensionali su carta. Questo articolo presenta un nuovo potente modello di intelligenza artificiale in grado di apprendere dalle forme 3D di molti tipi di molecole contemporaneamente — da piccoli composti farmaceutici a grandi proteine e i loro complessi — e di usare poi quella conoscenza per prevedere quanto intensamente interagiscono e quali potrebbero diventare futuri farmaci.
Una sola mappa per molti mondi molecolari
La maggior parte degli strumenti di IA attuali per la chimica sono specialisti: uno è addestrato solo su piccole molecole, un altro solo su proteine e un terzo solo sui loro complessi. Questa separazione spreca dati e rende difficile trasferire ciò che si impara in un campo a un altro. Gli autori costruiscono invece un unico modello «fondazione», chiamato Equivariant Pretrained Transformer (EPT), che apprende da un vasto insieme di strutture molecolari 3D tratte da più banche dati pubbliche. Trattando tutte queste strutture in un quadro condiviso, il modello può riconoscere schemi comuni nell’organizzazione e nell’interazione degli atomi, che si tratti di una semplice piccola molecola farmaceutica o di un intricato groviglio di catene proteiche. 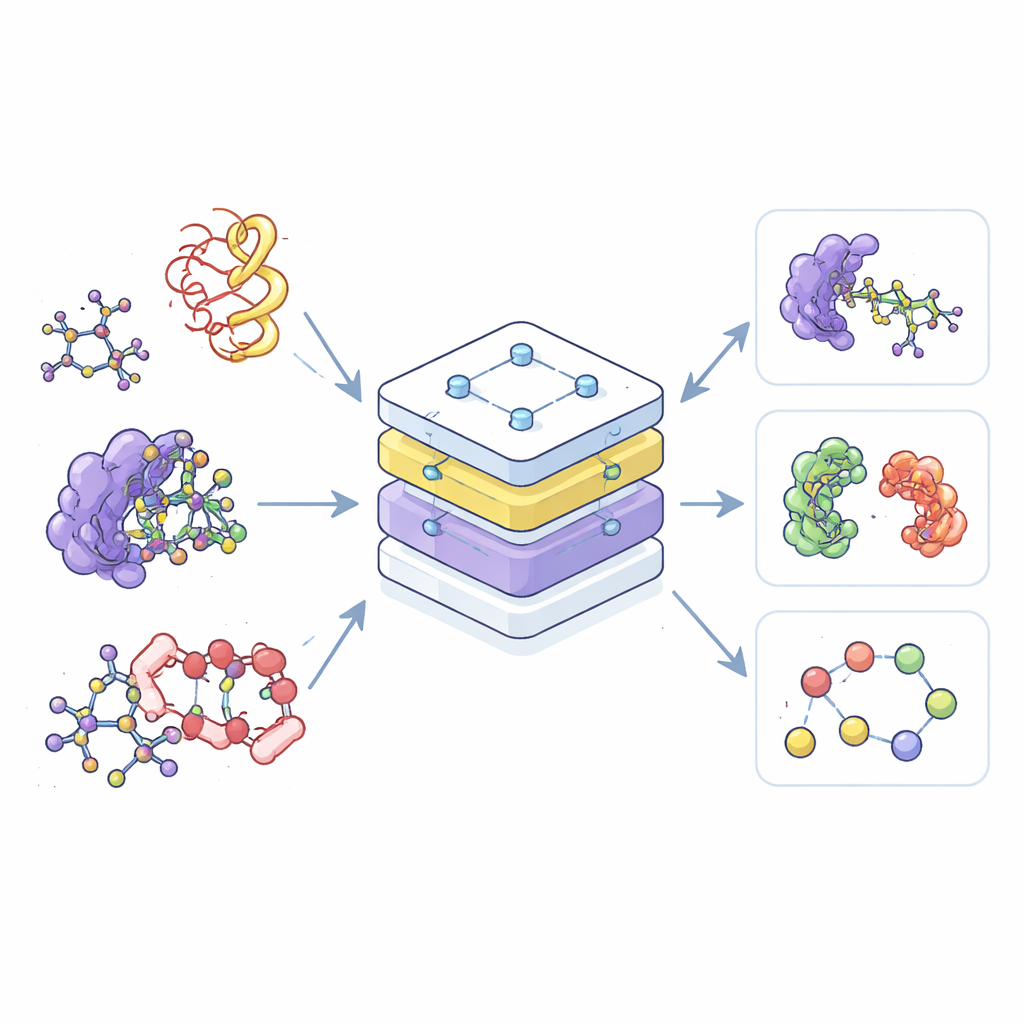
Spezzare le molecole in pezzi gestibili
Per affrontare l’enorme varietà e le diverse dimensioni dei sistemi molecolari, i ricercatori introducono l’idea dei «blocchi» — piccoli pezzi di atomi con significato chimico. Per le piccole molecole, un blocco raggruppa un atomo pesante con gli idrogeni a esso legati; per le proteine, ogni amminoacido diventa un blocco. Durante l’addestramento il modello vede sia gli atomi più dettagliati sia la struttura a blocchi più grossolana, permettendogli di collegare i dettagli chimici locali a forme 3D più ampie come la spina dorsale della proteina o le tasche di legame. Questa vista a blocchi crea anche un linguaggio comune che funziona attraverso tipi molecolari molto diversi, rendendo possibile a un unico modello comprenderli tutti.
Imparare riparando strutture rumorose
Invece di essere istruito con etichette esplicite del tipo «questa molecola è solubile» o «questa si lega tenacemente», EPT viene addestrato in modo self-supervised. Gli autori perturbano deliberatamente ogni blocco molecolare, spostandolo e ruotandolo a caso dalla sua posizione reale, e poi chiedono al modello di inferire le forze e le torsioni necessarie a ripristinare la struttura originale. Poiché l’addestramento rispetta le regole geometriche fondamentali — la molecola dovrebbe apparire la stessa se l’intero sistema viene ruotato o traslato nello spazio — il modello apprende un senso fisicamente coerente della forma 3D. Questo gioco di denoising insegna a EPT come gli atomi si tengono insieme all’interno e tra i blocchi e come cambiamenti sottili nella geometria influenzino la stabilità. 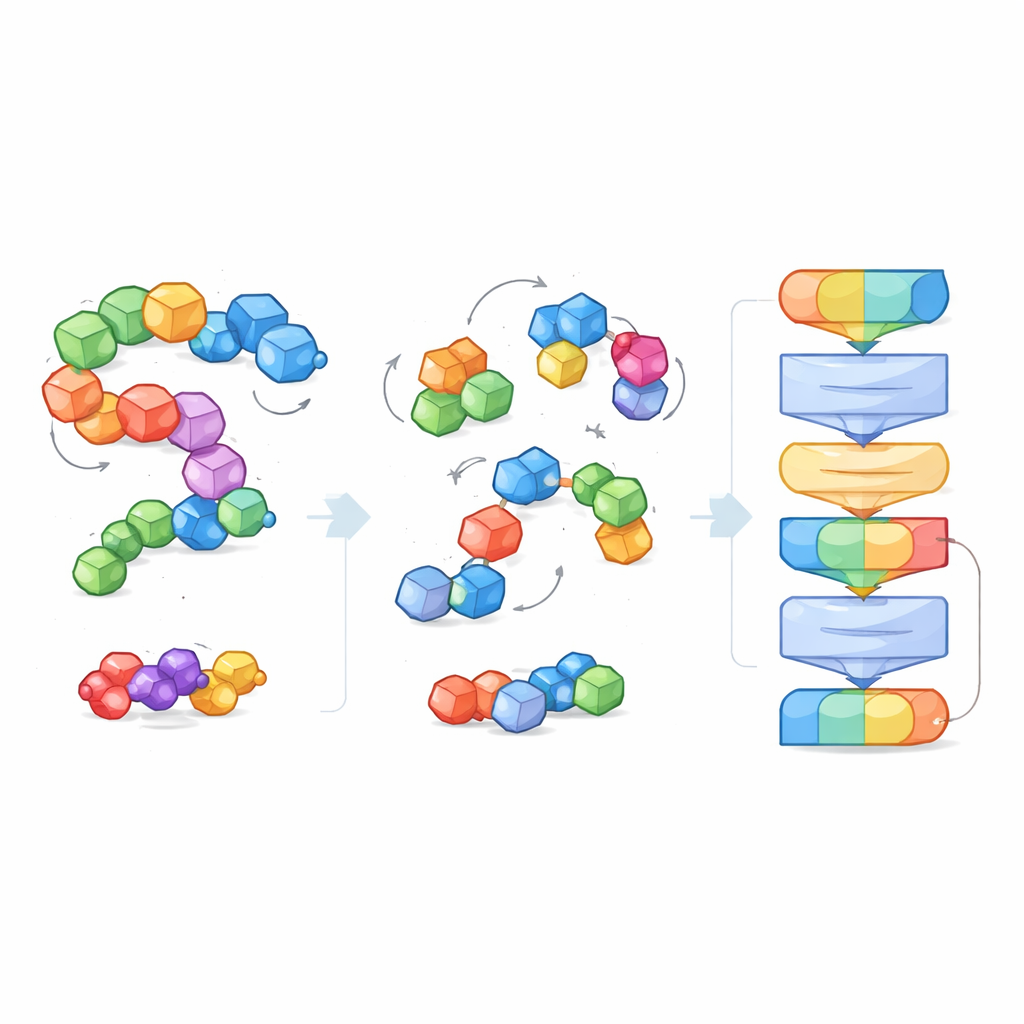
Mettere il modello alla prova
Dopo il preaddestramento su più di cinque milioni di strutture, EPT viene adattato (fine-tuned) per diversi compiti scientifici reali. Predice quanto fortemente una piccola molecola si leghi a una tasca proteica, come una singola mutazione in un’interfaccia proteica influenzi il legame e proprietà fisiche chiave di piccole molecole che interessano i chimici. Su benchmark diversi, il modello unificato eguaglia o supera i migliori strumenti esistenti, compresi quelli sviluppati appositamente per un solo dominio. Notevolmente, quando è addestrato su un tipo di dati, come le piccole molecole, risulta ancora utile per compiti apparentemente diversi, come la previsione del legame proteico, rivelando che ha catturato principi chimici ampiamente utili piuttosto che trucchi ristretti.
Alla ricerca di nuovi trattamenti per il COVID‑19
Gli autori dimostrano inoltre il valore pratico di EPT applicandolo a una sfida di riposizionamento di farmaci. Prima adattano il modello su complessi proteina–ligando e poi lo usano per classificare quasi 2.000 farmaci già approvati in base alla loro capacità prevista di legare la principale proteasi di SARS‑CoV‑2, un enzima chiave necessario al virus per replicarsi. Farmaci noti contro il COVID‑19 risalgono verso la parte alta della classifica e il modello mette in evidenza ulteriori candidati promettenti. Dodici molecole con punteggi elevati vengono esaminate più da vicino con simulazioni al computer e due — inclusa una non originariamente sviluppata per il COVID‑19 — mostrano un legame predetto particolarmente forte e sono confermate sperimentalmente come inibitori della proteasi virale a livelli micromolari.
Un passo verso un’IA molecolare generale
In termini semplici, questo lavoro dimostra che un unico modello di IA consapevole della geometria può apprendere una comprensione 3D condivisa di molti sistemi molecolari e poi usarla per rispondere a una vasta gamma di domande scientifiche. Organizzando le molecole in blocchi e addestrando il modello a «riparare» strutture distorte, gli autori creano uno strumento che non solo predice valori con maggiore accuratezza, ma può anche accelerare compiti come la scoperta di nuovi antivirali. EPT indica verso un futuro in cui sistemi di IA molecolare di uso generale aiutano chimici e biologi a esplorare lo spazio chimico in modo più efficiente, guidando esperimenti e accorciando il percorso dalla struttura atomica a terapie e materiali pratici.
Citazione: Jiao, R., Kong, X., Zhang, L. et al. An equivariant pretrained transformer for unified 3D molecular representation learning. Nat Commun 17, 2606 (2026). https://doi.org/10.1038/s41467-026-69185-7
Parole chiave: rappresentazione molecolare 3D, trasformatore equivariant, scoperta di farmaci, legame proteina–ligando, apprendimento self-supervised