Clear Sky Science · it
Scoprire la diversità dei PAM di Cas9 attraverso il mining metagenomico e il machine learning
Perché questo conta per il futuro dell’editing genico
CRISPR è diventato il simbolo dell’editing genico moderno, tuttavia una regola discreta continua a limitarne le applicazioni: ogni taglio nel DNA deve trovarsi accanto a una breve sequenza “permesso”. Questi brevi motivi, chiamati PAM, decidono dove l’enzima Cas9 popolare può e non può operare. Questo studio mostra come setacciare vasti volumi di DNA microbico, combinato con avanzati metodi di machine learning, possa rivelare un’enorme varietà nascosta di questi “permessi”. Questa nuova mappa potrebbe aprire molti più punti nel genoma umano a terapie precise e più sicure.
Regole nascoste che guidano i tagli CRISPR
Cas9 e enzimi correlati fanno parte di un sistema immunitario naturale presente in batteri e archea. Per evitare di tagliare il proprio DNA, questi microrganismi fanno sì che le proteine Cas cerchino un PAM—una brevissima sequenza di basi—accanto al sito bersaglio. Solo quando questo PAM è presente Cas9 srotola il DNA e permette al suo RNA guida di verificare la corrispondenza, innescando un taglio se tutto coincide. Il problema per la medicina è che i comunemente usati strumenti di laboratorio, come il Cas9 standard di Streptococcus pyogenes, riconoscono solo motivi PAM molto restrittivi. Se una mutazione responsabile di una malattia non possiede la sequenza adatta nelle vicinanze, gli strumenti attuali semplicemente non riescono a raggiungerla senza compromettere la precisione.
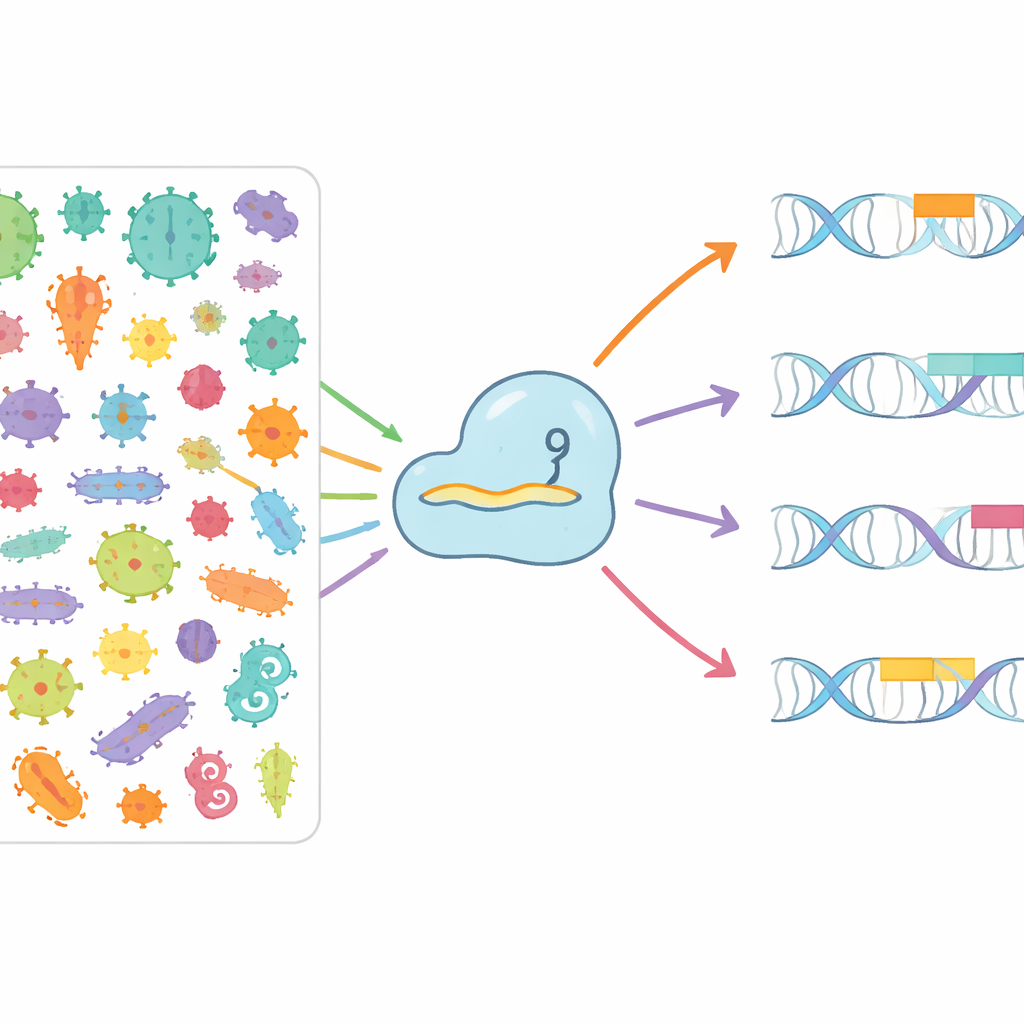
Minerare il mondo microbico per nuove opzioni
Gli autori si sono posti l’obiettivo di mappare sistematicamente come differenti proteine Cas9 riconoscano diversi PAM in natura. Hanno setacciato più di 3,8 milioni di genomi batterici e archeali e oltre 7,4 milioni di sequenze virali e di plasmidi che infettano o si spostano tra i microrganismi. Identificando gli array CRISPR, collegandoli ai geni Cas9 vicini e poi confrontando gli spacers di “memoria” immagazzinati con i virus e plasmidi invasori, hanno potuto vedere quali brevi motivi di DNA tendevano a fiancheggiare bersagli reali. Da questo hanno costruito CRISPR-PAMdb, un catalogo pubblico contenente 8003 gruppi di Cas9, ciascuno associato a un profilo PAM consenso, e organizzato in un albero evolutivo che evidenzia come enzimi Cas9 strettamente correlati tendano a condividere preferenze PAM simili pur mostrando una notevole diversità complessiva.
Quando i dati non bastano, lascia che il modello impari
Anche con questo enorme sondaggio, la maggior parte delle proteine Cas9 trovate non aveva sufficienti bersagli virali corrispondenti per leggere direttamente un PAM. Per colmare queste lacune, il team ha sviluppato un modello di machine learning chiamato CICERO. CICERO sfrutta un potente “modello linguistico” per proteine che ha appreso pattern generali delle sequenze di amminoacidi e lo affina per prevedere, per una data proteina Cas9, quanto è probabile che ciascuna base di DNA compaia in ciascuna delle dieci posizioni del PAM. Il modello è stato addestrato sui profili PAM di CRISPR-PAMdb e poi testato sia mediante cross-validation sia su 79 enzimi Cas9 i cui PAM erano stati misurati sperimentalmente, ottenendo una forte corrispondenza tra previsione e realtà.
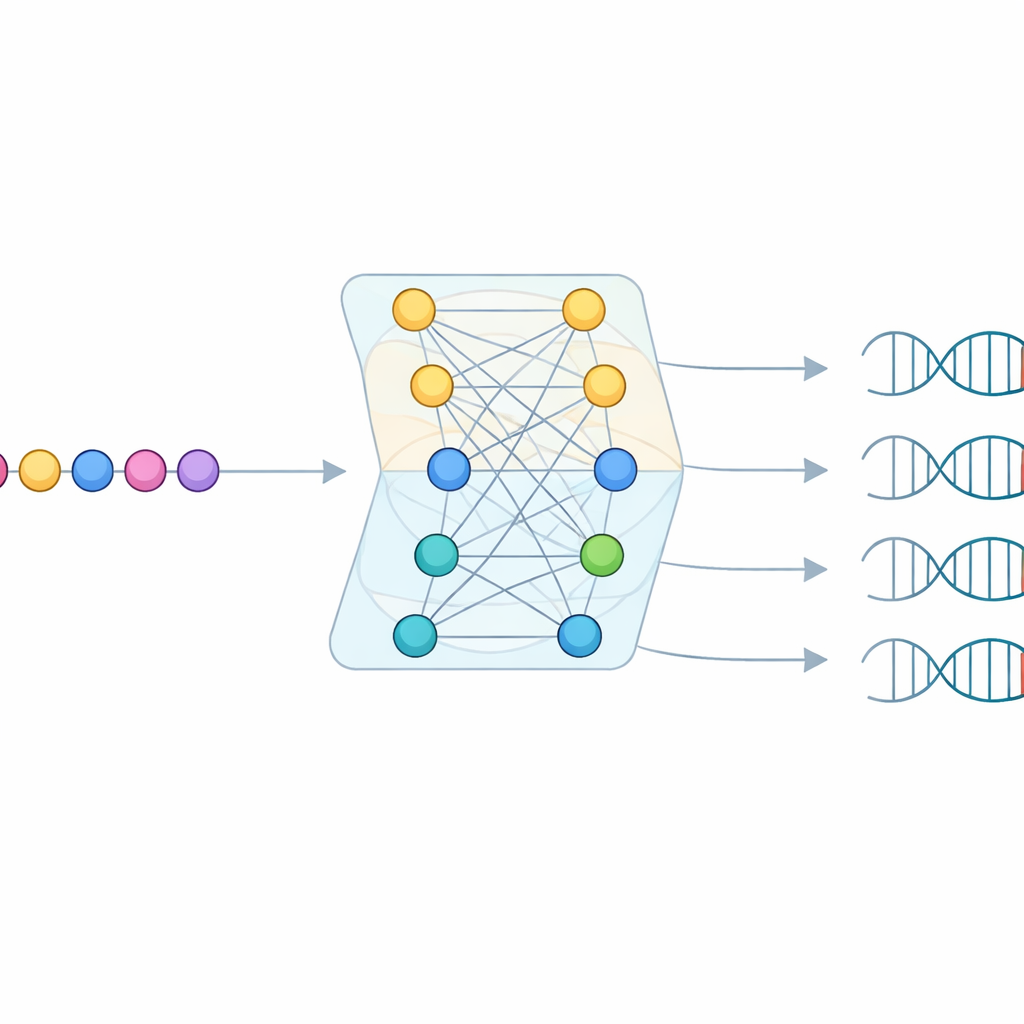
Sapere quanto essere fiduciosi
Una caratteristica chiave di CICERO è che non si limita a ipotizzare un PAM—stima anche quanto sia affidabile ciascuna previsione. Dopo aver imparato a prevedere i pattern PAM, i ricercatori hanno addestrato una seconda rete, leggera, che prende la stessa sequenza di Cas9 e impara a prevedere quanto sarà accurata la previsione del PAM. Punteggi di fiducia più alti si correlavano fortemente con una maggiore accuratezza nel mondo reale. Utilizzando questo filtro di confidenza, il team ha esteso le annotazioni PAM a più di 50.000 proteine Cas9 aggiuntive, con oltre 17.000 previsioni classificate ad alta fiducia. Questo amplia notevolmente il repertorio di varianti Cas9 con regole di targeting ragionevolmente ben comprese.
Cosa significa per il trattamento delle malattie genetiche
Per mostrare l’importanza di queste nuove risorse, gli autori hanno esaminato decine di migliaia di mutazioni legate a malattie nel database ClinVar che in linea di principio potrebbero essere corrette usando editor di basi—strumenti che cambiano una singola base del DNA senza tagliare entrambi i filamenti. Hanno scoperto che l’enzima Cas9 standard può raggiungere solo circa la metà di questi siti a causa delle sue esigenze stringenti sul PAM. Quando hanno preso in considerazione i parenti di Cas9 presenti in CRISPR-PAMdb e le previsioni ad alta confidenza di CICERO che riconoscono un insieme più ampio ma ancora specifico di sequenze vicine, quasi tutte queste mutazioni sono diventate teoricamente raggiungibili senza allentare il targeting a tal punto da perdere precisione.
Un toolkit più ampio per la chirurgia del DNA precisa
In termini semplici, questo lavoro costruisce due cose: una gigantesca mappa pubblica che collega migliaia di proteine Cas9 naturali ai brevi motivi del DNA che preferiscono, e una guida basata su AI che può prevedere queste preferenze per molte altre proteine semplicemente dalle loro sequenze. Insieme, trasformano il mondo microbico in un ricco catalogo di parti per i futuri editor genici. Man mano che i ricercatori perfezioneranno e testeranno queste varianti Cas9 in laboratorio, i clinici potrebbero ottenere strumenti più sicuri e versatili in grado di raggiungere mutazioni causative di malattie precedentemente inaccessibili, avvicinando la chirurgia genomica veramente precisa a una realtà concreta.
Citazione: Fang, T., Bogensperger, L., Feer, L. et al. Uncovering Cas9 PAM diversity through metagenomic mining and machine learning. Nat Commun 17, 2510 (2026). https://doi.org/10.1038/s41467-026-69098-5
Parole chiave: CRISPR-Cas9, diversità PAM, metagenomica, machine learning, editing del genoma