Clear Sky Science · it
Predizione della somiglianza chimica tra spettri di massa tandem in modalità di ionizzazione incrociata in metabolomica
Perché collegare i punti chimici è importante
Ogni sorso di caffè, respiro d’aria o cura medica lascia piccole tracce chimiche nei nostri corpi. Gli strumenti moderni possono rilevare migliaia di queste molecole contemporaneamente, ma tradurre quei segnali in informazioni biologiche rimane sorprendentemente difficile. Questo studio presenta MS2DeepScore 2.0, uno strumento di machine learning che aiuta gli scienziati a comprendere come queste molecole siano correlate, anche quando i segnali vengono registrati in modi molto diversi. Così facendo, promette interpretazioni più rapide e più complete di miscele chimiche complesse in medicina, nutrizione e ricerca ambientale.
Due modi di osservare la stessa molecola
La spettrometria di massa è una tecnica fondamentale che pesa e frammenta le molecole per rivelarne l’identità. Negli esperimenti di routine, gli scienziati spesso misurano lo stesso campione in due modalità: una che favorisce molecole cariche positivamente e una che favorisce quelle cariche negativamente. Ciascuna modalità produce un proprio “codice a barre” caratteristico di frammenti. Anche quando entrambe le misure provengono dalla stessa molecola, i modelli risultanti possono sembrare così diversi che i metodi di confronto tradizionali falliscono. Di conseguenza, i ricercatori di solito analizzano le due modalità separatamente, costruendo due mappe disconnesse del campione e rischiando di perdere relazioni chimiche importanti.
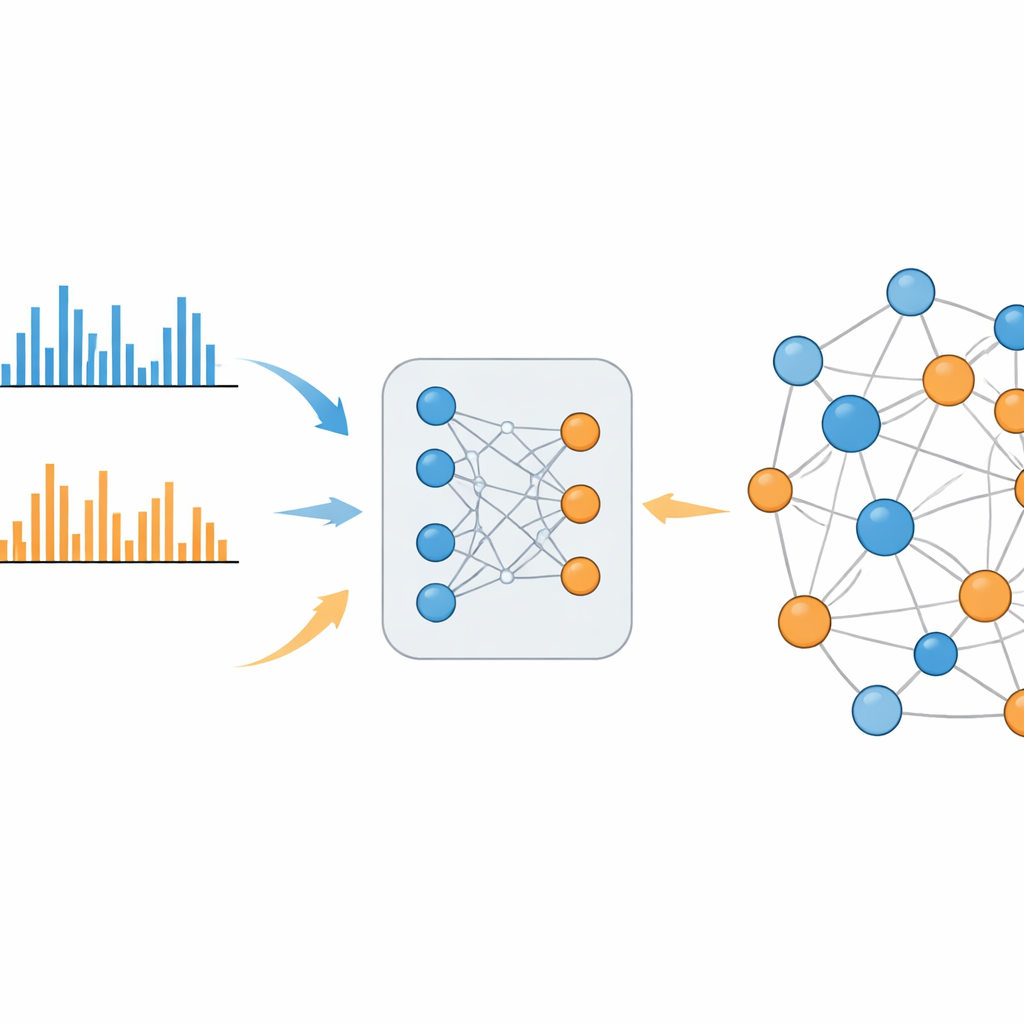
Un sistema di apprendimento che colma il divario
MS2DeepScore 2.0 affronta questa divisione apprendendo la somiglianza chimica direttamente da ampie librerie di spettri noti. Il modello si basa su un’architettura a rete neurale gemella che trasforma ogni spettro di frammentazione in un’impronta composta da 500 numeri, chiamata embedding. Durante l’addestramento, il sistema vede centinaia di migliaia di esempi provenienti sia dalla modalità positiva sia da quella negativa, insieme alle informazioni su quanto sono effettivamente simili le molecole sottostanti. Si aggiusta in modo che spettri di molecole correlate finiscano per avere embedding simili, indipendentemente dal fatto che siano stati misurati nella stessa modalità o in modalità opposte. La nuova versione va oltre il predecessore inserendo informazioni aggiuntive, come la massa della molecola originale e la modalità di ionizzazione utilizzata, e utilizzando uno schema di campionamento bilanciato in modo che relazioni chimiche rare ma informative non vengano sommerse da quelle comuni e poco informative.
Da segnali dispersi a mappe unificate
Una volta addestrato, MS2DeepScore 2.0 può stimare quanto siano chimicamente simili due spettri qualsiasi, incluse coppie positive vs negative. Gli autori dimostrano che queste previsioni si correlano bene con misure consolidate di somiglianza strutturale, non solo all’interno di ciascuna modalità ma anche attraverso le modalità. Usando dati reali provenienti da urine umane, plasma sanguigno umano e una pianta selvatica commestibile, costruiscono “network molecolari” in cui ogni spettro è un nodo e una forte somiglianza predetta crea una connessione. A differenza degli approcci precedenti, queste reti mescolano naturalmente dati delle modalità positiva e negativa in mappe uniche e coerenti. Cluster curati da esperti includono, per esempio, gruppi di molecole correlate alla caffeina nelle urine che sono collegate attraverso le modalità di ionizzazione e corrispondono a vie metaboliche note.
Vedere il paesaggio chimico a colpo d’occhio
I network molecolari sono potenti ma possono diventare aggrovigliati se si includono troppe connessioni deboli. Per evitare ciò, gli autori usano gli embedding di MS2DeepScore direttamente come coordinate in un layout bidimensionale creato con una tecnica chiamata UMAP. Ogni punto in questa mappa rappresenta uno spettro, e i punti vicini corrispondono a molecole che il modello considera chimicamente simili. Spettri in modalità positiva e negativa dello stesso composto, che a occhio appaiono molto diversi, spesso finiscono fianco a fianco in questo spazio di embedding. Il gruppo addestra inoltre un modello aggiuntivo che esamina ogni embedding e ne stima l’affidabilità, segnalando spettri rumorosi, incompleti o diversi da qualsiasi cosa vista durante l’addestramento. Rimuovere questi punti di bassa qualità migliora l’accuratezza complessiva e rende le visualizzazioni più affidabili.
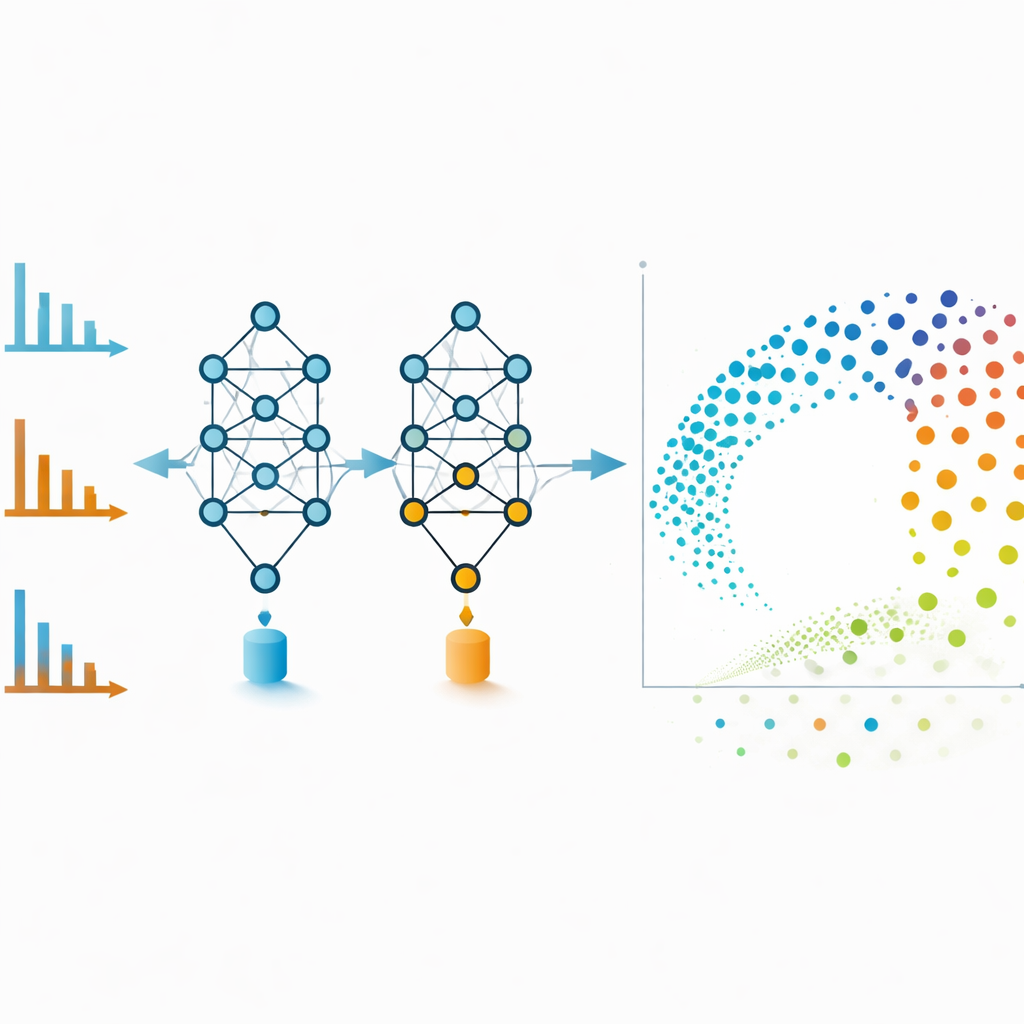
Portare strumenti avanzati nei laboratori di tutti i giorni
Per garantire che questa tecnologia sia utilizzabile anche da chi non è esperto di programmazione, gli autori hanno integrato MS2DeepScore 2.0 in software di spettrometria di massa popolari e gratuiti. Con questa integrazione, i ricercatori possono rilevare feature, costruire network molecolari che ignorano i confini tra modalità di ionizzazione ed esplorare lo spazio chimico risultante tramite dashboard interattive. Il codice, i modelli addestrati e i dataset d’esempio sono condivisi apertamente, e il sistema può essere riaddestrato o perfezionato per classi chimiche specializzate.
Cosa significa per le scoperte future
Per i non specialisti, il messaggio chiave è che MS2DeepScore 2.0 aiuta a trasformare misurazioni frammentate e dipendenti dalla modalità in un’unica immagine più comprensibile delle molecole presenti in un campione. Collegando in modo affidabile segnali che prima vivevano in mondi analitici separati, il metodo consente agli scienziati di sfruttare librerie di riferimento molto più grandi, confrontare i campioni in modo più completo e concentrare l’attenzione su cluster significativi di composti correlati. Questo incrocio di dati dovrebbe accelerare l’identificazione di biomarcatori, nutrienti, prodotti naturali e inquinanti, approfondendo in ultima analisi la nostra comprensione di come la chimica influenzi la salute e l’ambiente.
Citazione: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Parole chiave: metabolomica, spettrometria di massa, apprendimento automatico, networking molecolare, somiglianza chimica