Clear Sky Science · it
Un modello polimerico informato sperimentalmente rivela l’organizzazione ad alta risoluzione dei loci genici
Come il ripiegamento del DNA plasma l’identità cellulare
Ogni cellula del corpo contiene sostanzialmente lo stesso DNA, eppure le cellule cerebrali, cutanee e le cellule staminali si comportano in modo molto diverso. Una ragione chiave è il modo in cui quel DNA è ripiegato e impacchettato all’interno del nucleo. Questo studio introduce un nuovo metodo per “vedere” tale ripiegamento in dettagli notevoli, collegando l’organizzazione fisica del DNA al fatto che geni importanti siano attivati o silenziati. Combinando esperimenti con simulazioni al computer basate sulla fisica, gli autori rivelano ammassi nascosti di materiale genetico che sembrano agire come unità fondamentali dell’organizzazione del genoma.
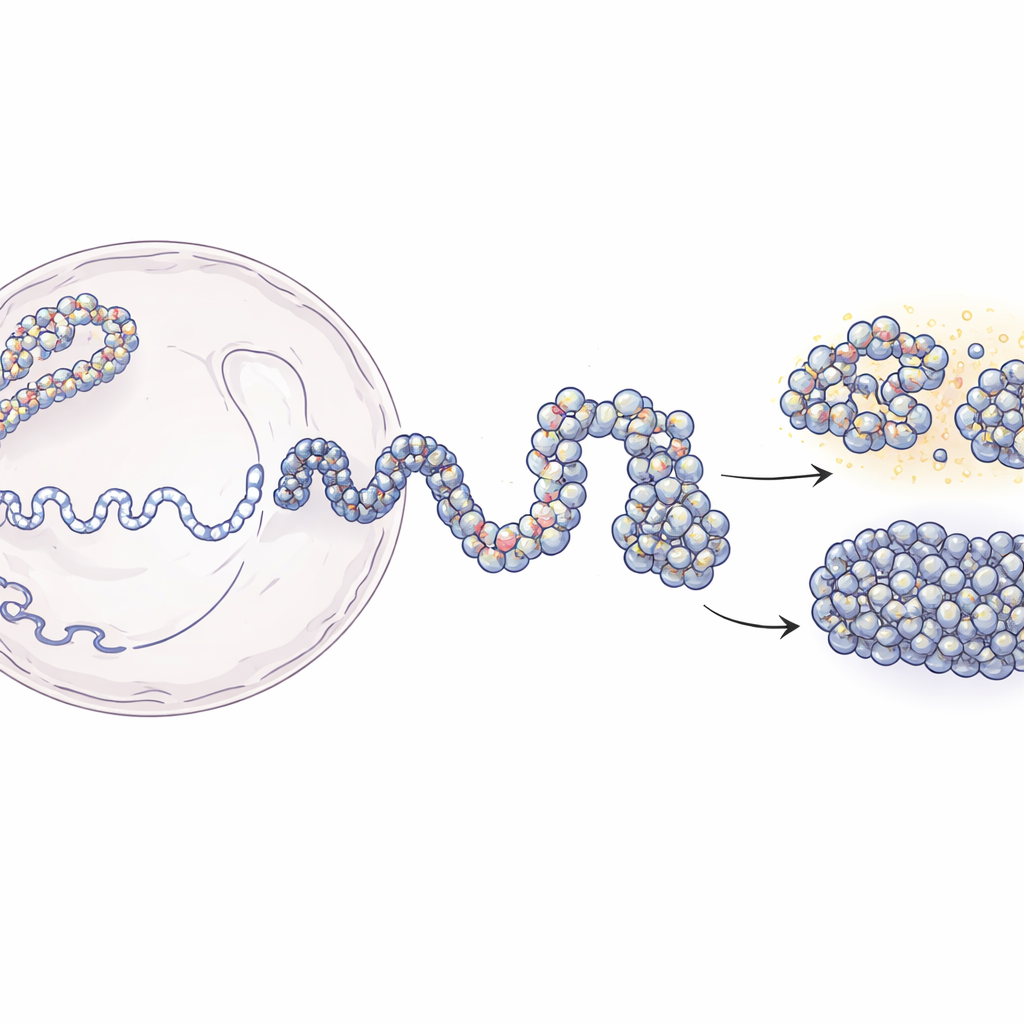
Dai lunghi fili di DNA alle mappe 3D del genoma
Nel nucleo, il DNA è avvolto attorno a spool proteici chiamati nucleosomi, formando una struttura a perline nota come cromatina. Tecniche moderne come Hi-C e Micro-C ci dicono quali porzioni di DNA sono vicine nello spazio 3D, ma di solito forniscono immagini sfocate mediate su popolazioni di cellule. Al contrario, esperimenti che localizzano singoli nucleosomi offrono dettagli locali nitidi ma poco senso della struttura globale. Questo lavoro colma quel divario. Gli autori partono da mappe di contatto a bassa risoluzione che riportano la frequenza con cui segmenti distanti di DNA si toccano, quindi le combinano con mappe sperimentali delle posizioni dei nucleosomi. Usando principi della fisica dei polimeri, costruiscono insiemi 3D simulati di cromatina che corrispondono ai dati sperimentali ma risolvono strutture fino a poche decine di basi di DNA.
Una strategia in due passi per ricostruire la cromatina
L’approccio di modellizzazione si sviluppa in due fasi principali. Prima, il gruppo usa dati Hi-C per generare molte possibili forme su larga scala di un tratto di DNA di 200.000 basi, trattando la cromatina come una catena flessibile in cui segmenti da 5.000 basi sono guidati dolcemente a formare o evitare contatti come osservato negli esperimenti. Queste strutture grossolane catturano il pattern generale di ripiegamento che le proteine cellulari contribuiscono a creare. Nella seconda fase, ogni grande “perla” viene sostituita da una catena molto più fine composta da singoli nucleosomi e dai brevi linker di DNA tra di essi. Le posizioni di questi nucleosomi provengono da un metodo di mappatura basato su enzimi (MNase-seq) che rivela dove tipicamente si collocano lungo il genoma. Le catene a risoluzione fine sono quindi lasciate ripiegare pur rispettando l’architettura più ampia. Quando i ricercatori “sfocano” i loro modelli ad alta risoluzione riportandoli alle risoluzioni sperimentali, riproducono con alta accuratezza sia le mappe di contatto Hi-C sia Micro-C.
Scoperta degli aggregati di nucleosomi come unità strutturali
Quando gli autori hanno ingrandito le loro strutture simulate è emerso un pattern sorprendente: i nucleosomi non erano disposti uniformemente, ma si raccoglievano in ammassi irregolari, che gli autori chiamano aggregati di nucleosomi. Questi aggregati assomigliano alle strutture raggrumate osservate in immagini da microscopio a super-risoluzione di cellule reali. Analizzando migliaia di istantanee simulate, il team ha mostrato che questi aggregati sono allungati, non sferici, e tipicamente contengono diversi nucleosomi ravvicinati. Crucialmente, i contatti all’interno di questi aggregati corrispondono strettamente ai blocchi di interazione simili a domini osservati nei dati sperimentali, indicando che gli aggregati non sono incidenti casuali ma unità 3D fondamentali dell’organizzazione della cromatina. Le simulazioni predicono persino confini di dominio sottili aggiuntivi, difficili da rilevare sperimentalmente, suggerendo che questo modello fisico possa scoprire caratteristiche a scala fine nascoste nei dati rumorosi.
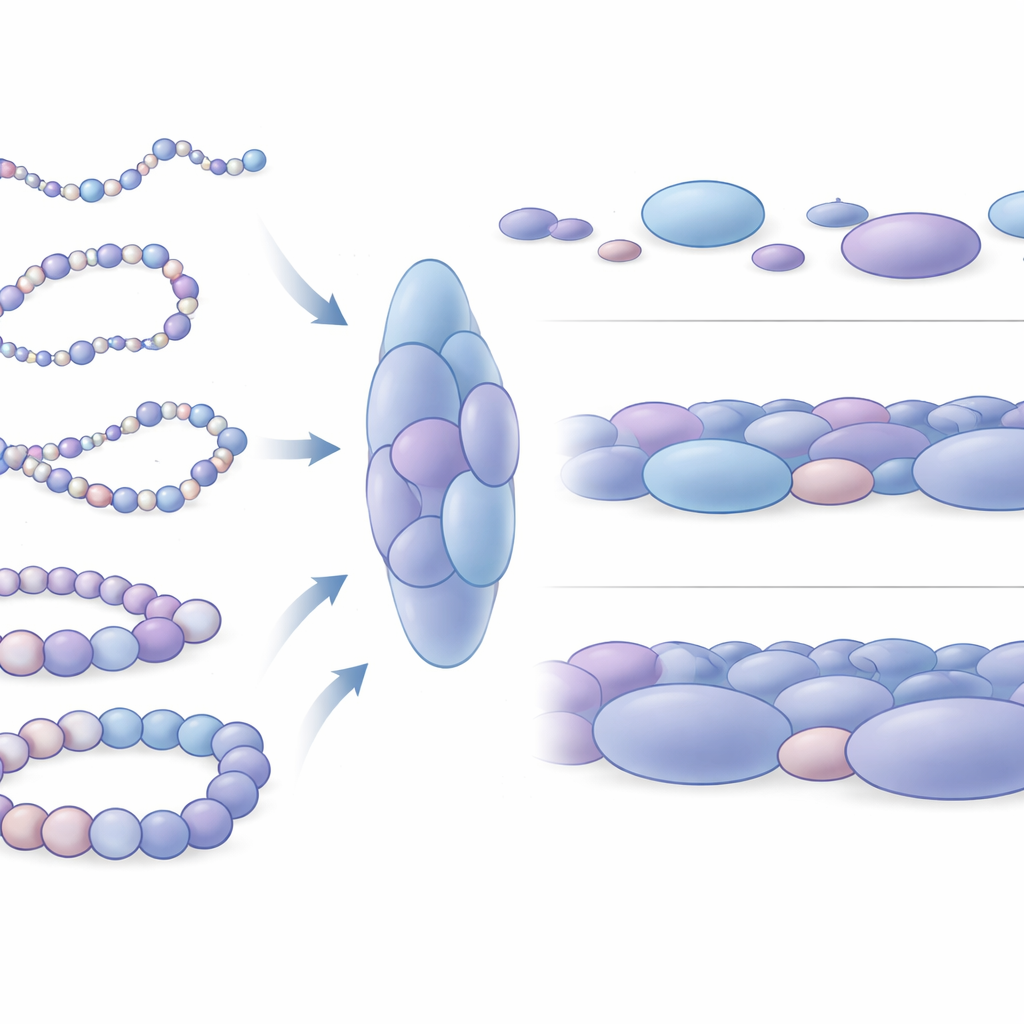
Come le differenze di impacchettamento riflettono l’attività genica
I ricercatori hanno poi indagato come questi aggregati differiscano attorno a geni attivi rispetto a quelli silenti. Si sono concentrati su quattro tratti ben studiati di DNA umano, inclusi due geni che aiutano a mantenere le cellule staminali in uno stato flessibile e indifferenziato (Nanog e Lin28A) e due geni di controllo dello sviluppo (HoxB4 e HoxA13) che sono spenti nelle stesse cellule. Attorno ai geni inattivi, gli aggregati erano mediamente più grandi e più compatti, con nucleosomi che formavano disposizioni locali più chiuse. Al contrario, gli aggregati vicino ai geni attivi erano più piccoli, un po’ più lassi e più variabili. Su scala più ampia, il DNA attorno ai geni attivi campionava molte più forme distinte ed era meccanicamente più flessibile, mentre le regioni intorno ai geni silenti si comportavano come segmenti di cromatina più rigidi. Questa differenza meccanica probabilmente influenza quanto facilmente elementi regolatori distanti del DNA possono incontrarsi e cooperare con gli interruttori genici.
Perché questo è importante per comprendere il controllo genico
Nel complesso, i risultati dipingono un quadro in cui il genoma è costruito da aggregati dinamici di nucleosomi la cui dimensione, forma e spaziatura aiutano a determinare se i geni vicini sono accessibili o sigillati. Il nuovo modello collega dati sperimentali di contatto, mappe dei nucleosomi e principi fisici in un unico quadro che spiega come i geni delle cellule staminali possano restare flessibili e interattivi mentre i geni dello sviluppo rimangono sequestrati in quartieri più compatti e rigidi. Per i non specialisti, l’idea chiave è che l’attività genica non è governata solo dalla sequenza del DNA; dipende anche da come quel DNA è ripiegato in strutture tridimensionali. Rivelando gli aggregati di nucleosomi come mattoni fondamentali di tale ripiegamento, questo lavoro offre una via potente per collegare l’architettura microscopica del genoma a processi su larga scala come lo sviluppo, l’identità cellulare e la malattia.
Citazione: Mittal, R., Heermann, D.W. & Bhattacherjee, A. An experimentally-informed polymer model reveals high resolution organization of genomic loci. Nat Commun 17, 2338 (2026). https://doi.org/10.1038/s41467-026-68928-w
Parole chiave: ripiegamento della cromatina, aggregati di nucleosomi, organizzazione 3D del genoma, regolazione genica, modellizzazione polimerica