Clear Sky Science · it
Previsione affidabile dei numeri Enzyme Commission usando un trasformatore gerarchico e interpretabile
Perché è importante prevedere il ruolo degli enzimi
Ogni cellula vivente funziona grazie a innumerevoli minuscole macchine chimiche chiamate enzimi. Ogni enzima svolge un “compito” specifico, codificato in un numero Enzyme Commission (EC), un codice in quattro parti un po’ come un indirizzo postale. Assegnare correttamente i numeri EC è cruciale per comprendere il metabolismo, progettare nuovi farmaci, ingegnerizzare microrganismi per produrre combustibili o alternative alla plastica e monitorare come gli ecosistemi trasformano le sostanze chimiche. Tuttavia, gli esperimenti per determinare le funzioni enzimatiche sono lenti e costosi. Questo studio presenta HIT-EC, un nuovo modello di intelligenza artificiale in grado di prevedere in modo affidabile i numeri EC a partire dalle sequenze proteiche offrendo anche spiegazioni sul perché di ciascuna previsione.
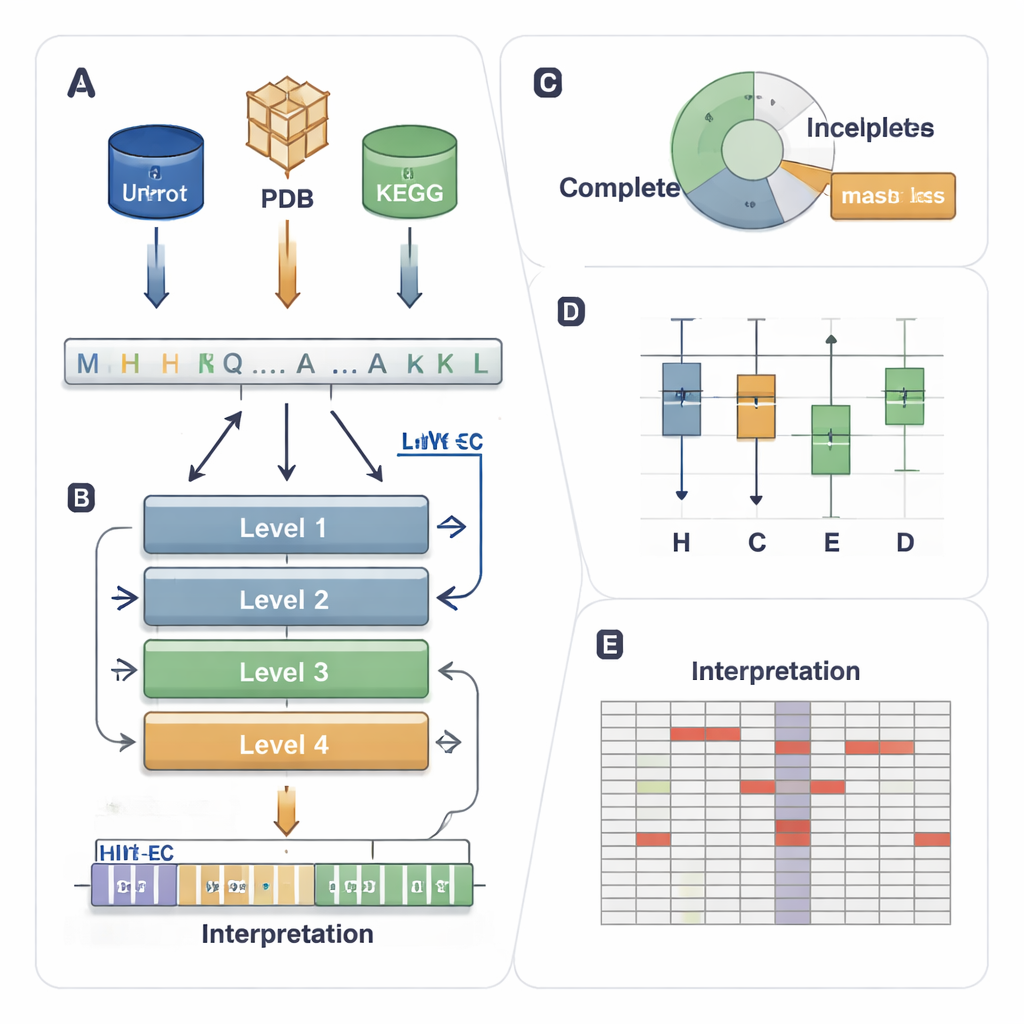
Un sistema tipo codice postale per i compiti enzimatici
Il sistema EC assegna a ogni enzima un codice a quattro livelli come 1.1.1.37. La prima cifra indica una classe ampia (per esempio enzimi che trasferiscono elettroni o gruppi), mentre le cifre successive descrivono dettagli di reazione più specifici. Questa gerarchia è potente ma crea un problema di previsione impegnativo: un modello deve indovinare correttamente tutti e quattro i livelli tra migliaia di codici possibili, anche quando alcuni enzimi sono rari o parzialmente annotati nei database (per esempio 3.5.-.-, dove mancano i livelli dettagliati). I metodi informatici esistenti usano struttura 3D, similarità di sequenza o deep learning, ma tendono a arrancare con enzimi poco comuni, ignorano dati parzialmente etichettati e si comportano tipicamente come “scatole nere” che offrono scarsa comprensione del motivo delle decisioni.
Un’IA a quattro piani che segue la scala EC
HIT-EC (Hierarchical Interpretable Transformer for EC prediction) è progettato per rispecchiare la gerarchia a quattro livelli dell’EC. Prende una sequenza proteica grezza e la fa passare attraverso quattro livelli transformer, ognuno focalizzato su un livello EC. Flussi locali collegano ogni livello al precedente, garantendo che una decisione fine (la quarta cifra) sia coerente con quelle più ampie (prima e seconda cifra). In parallelo, un flusso globale mantiene il contesto completo della sequenza visibile a ogni passo. Il modello può inoltre essere addestrato su sequenze con etichette incomplete, usando una “loss mascherata” che semplicemente ignora i livelli EC mancanti invece di scartare la sequenza. Questo permette a HIT-EC di imparare dalla grande frazione di proteine nei database curati che sono solo parzialmente annotate.
Prestazioni superiori in accuratezza e velocità
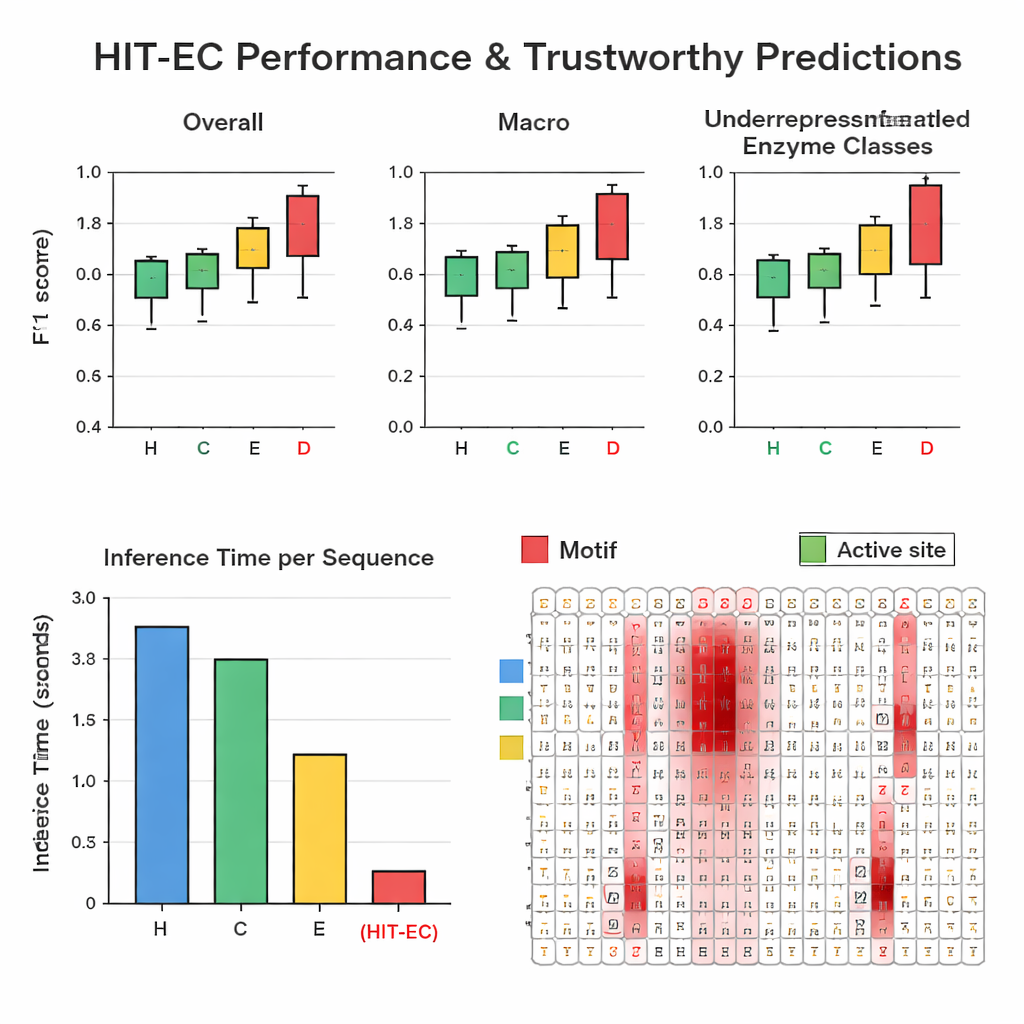
Gli autori hanno assemblato un ampio dataset accuratamente filtrato di circa 200.000 enzimi con 1.938 diversi numeri EC provenienti da Swiss-Prot e Protein Data Bank. In test ripetuti con hold-out, HIT-EC ha battuto tre metodi di riferimento (CLEAN, ECPICK e DeepECtransformer) sia su F1-score complessivi sia su F1-score per classe, che misurano l’equilibrio tra veri positivi e falsi allarmi. Si è mostrato particolarmente efficace sui codici EC sottorappresentati con 25 o meno esempi noti, dove i metodi precedenti spesso falliscono. HIT-EC ha anche generalizzato bene su enzimi nuovi aggiunti a Swiss-Prot dopo l’addestramento e su genomi completi di batteri diversi, incluse ceppi ben studiati di Escherichia coli, Bacillus subtilis e Mycobacterium tuberculosis. Nonostante la sua sofisticazione, il modello è altamente efficiente: su una GPU standard ha processato una proteina in circa 38 millisecondi—decine di volte più veloce di alcuni concorrenti che dipendono da ricerche di similarità più lente o da ensemble di molti modelli.
Mostrare cosa il modello “guarda”
Per rendere le sue previsioni affidabili, HIT-EC è progettato per mostrare quali amminoacidi nella sequenza hanno influenzato ogni decisione a livello EC. Gli autori hanno sviluppato un percorso di interpretazione che combina pesi di attenzione con informazioni sui gradienti per valutare l’importanza di ciascuna posizione. Hanno validato questi punteggi su famiglie enzimatiche ben caratterizzate. Per esempio, in una famiglia di citocromo P450 (CYP106A2), HIT-EC ha evidenziato motivi funzionali noti come le regioni di legame dell’ossigeno e dell’eme, e ha identificato un sottile motivo EXXR che un modello di riferimento non aveva colto. Per rappresentanti classici di ogni classe EC di primo livello—come alcol deidrogenasi, esochinasi e anidrasi carbonica—i punteggi di rilevanza del modello hanno evidenziato i motivi caratteristici dei manuali e i siti di legame del substrato. Queste interpretazioni forniscono “evidenze” biochimiche che il modello basa le sue decisioni su caratteristiche significative, e non su correlazioni accidentali.
Orientare il lavoro su enzimi rari ed emergenti
Il gruppo ha ulteriormente testato HIT-EC su due enzimi poco studiati importanti per la bonifica ambientale: un citocromo P450 coinvolto nella degradazione di inquinanti aromatici e un’idrolasi degradante PET da Streptomyces che aiuta a digerire molecole correlate alla plastica. Entrambi gli enzimi erano stati caratterizzati sperimentalmente ma mancavano di assegnazioni EC ufficiali. HIT-EC ha previsto correttamente i numeri EC attesi e ha evidenziato motivi e residui catalitici che corrispondono a quanto noto da studi strutturali e biochimici. Complessivamente, il lavoro mostra che HIT-EC può non solo assegnare numeri EC in modo più accurato e rapido rispetto agli strumenti attuali, specialmente per funzioni rare, ma anche spiegare perché un particolare enzima è ritenuto svolgere un dato compito chimico. Questa combinazione di prestazioni e interpretabilità lo rende un motore promettente per un’annotazione su larga scala, affidabile, di enzimi in genomica, biotecnologia e ricerca ambientale.
Citazione: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
Parole chiave: predizione della funzione degli enzimi, deep learning in biologia, modelli transformer, annotazione delle proteine, enzimi per biorisanamento