Clear Sky Science · it
Un’attivazione alternativa di EGFR dalla mutazione R252C derivata da paziente promuove la progressione del cancro
Quando le antenne cellulari diventano ribelli
Perché alcuni tumori continuano a crescere nonostante cicli di chemioterapia e le più moderne immunoterapie? Questo studio segue un paziente con tumori sia al polmone sia al cervello e ricostruisce la malattia a partire da una piccola alterazione in un’importante “antenna” di superficie cellulare chiamata EGFR. Svelando come questa singola mutazione riorienta i segnali di crescita, i ricercatori non solo spiegano l’aggressività del cancro del paziente, ma mostrano anche come un farmaco già esistente, l’afatinib, possa contenerla.
Una mutazione rara con grandi conseguenze
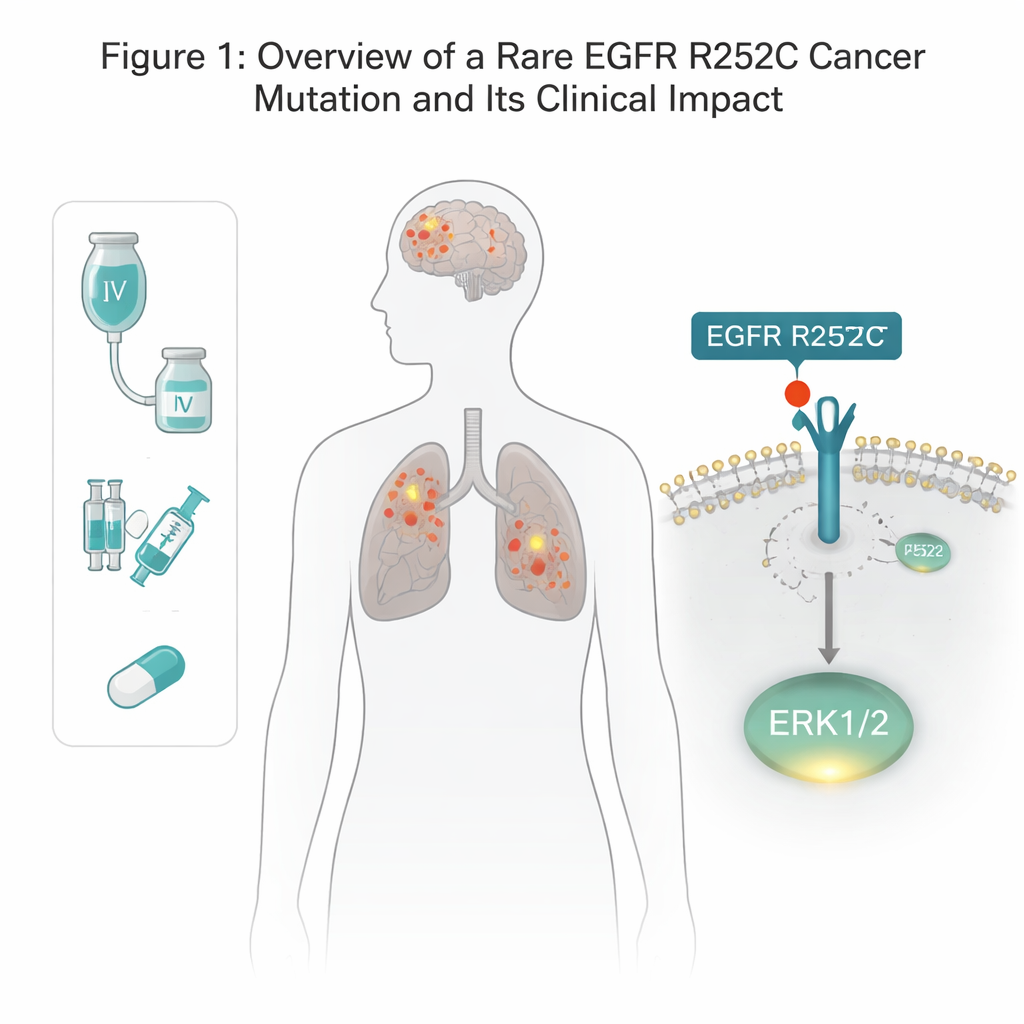
EGFR è un recettore che attraversa la membrana cellulare e aiuta le cellule a rispondere a segnali di crescita. Molti tumori polmonari e cerebrali presentano mutazioni in EGFR, ma la maggior parte delle alterazioni note si colloca all’interno della cellula, nella porzione che funziona come interruttore enzimatico. Qui, il gruppo ha scoperto una variazione insolita sulla parte esterna di EGFR, nella regione che normalmente lega i fattori di crescita. In un paziente con tumore al polmone e glioma è stato trovato che un singolo amminoacido alla posizione 252 era stato scambiato: dall’arginina alla cisteina — denominata EGFR R252C. L’analisi dei database sul cancro ha mostrato questa mutazione in una piccola frazione di pazienti con glioma e quasi mai nei tumori polmonari, suggerendo che è rara ma reale. Usando strumenti di editing genetico, gli autori hanno ricreato esattamente questa mutazione in diverse linee cellulari tumorali umane di cervello e polmone per testarne gli effetti.
Una nuova scorciatoia verso i segnali di crescita
Normalmente EGFR deve associarsi a un’altra copia e poi fosforilare la propria coda interna per attivare le vie di crescita a valle. Sorprendentemente, la versione R252C non mostrava questa fosforilazione tipica. Al contrario, le cellule che esprimevano EGFR R252C attivavano in modo molto più marcato un controllore di crescita specifico, ERK1/2, mentre altre vie classiche di EGFR — come AKT e STAT3 — rimanevano in gran parte invariate. Bloccare ERK1/2 con un inibitore dedicato annullava il vantaggio di crescita delle cellule R252C, dimostrando che ERK1/2 è il principale motore della capacità tumorale di questo mutante.
Bloccando il recettore in una conformazione sempre attiva
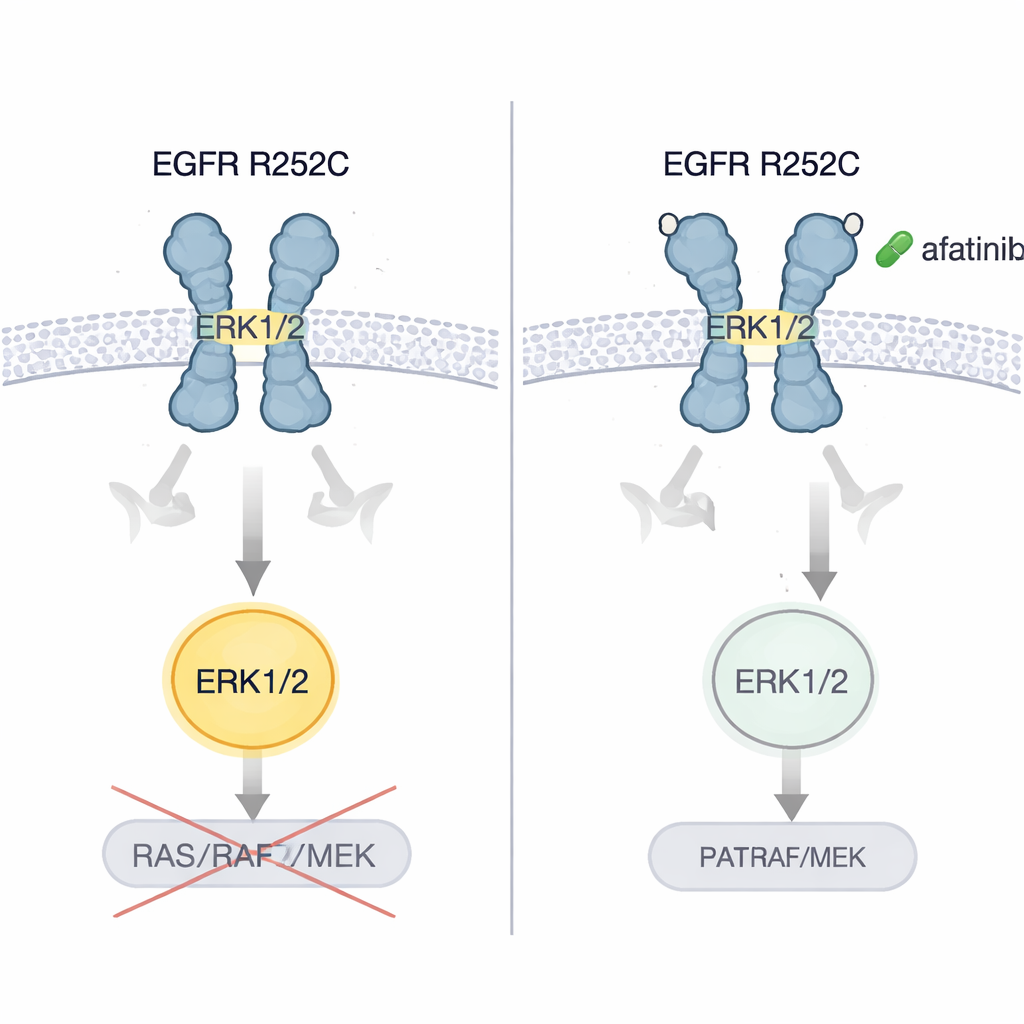
Per capire come una modifica esterna potesse scatenare un’attivazione così selettiva, i ricercatori hanno combinato saggi biochimici con simulazioni al computer. Lo scambio R252C introduce una nuova cisteina nella porzione esterna di EGFR. Due di questi mutanti possono formare un ponte disolfuro — una specie di graffetta molecolare — tra le loro residui C252, saldandosi in una coppia stabile. La modellizzazione strutturale ha mostrato che questo legame costringe l’esterno del recettore in un allineamento sfalsato a forma di “V” che imita da vicino la forma attiva legata al ligando, anche in assenza di fattore di crescita. Questo allineamento si propaga attraverso i segmenti transmembrana e quelli immediatamente interni, torcendo i domini enzimatici interni in una disposizione insolita: i siti attivi sono orientati verso l’interno della cellula ma distanziati troppo per fosforilarsi efficacemente a vicenda. Invece, questa conformazione crea una superficie di ancoraggio efficace per ERK1/2, permettendo a EGFR R252C di fosforilare ERK1/2 direttamente e bypassare la consueta cascata RAS–RAF–MEK.
Dai modelli murini a un singolo paziente
Gli autori hanno mostrato che le cellule tumorali cerebrali e polmonari che portavano EGFR R252C crescevano più rapidamente in coltura e formavano tumori più grandi e aggressivi quando impiantate nei topi, rispetto a cellule con EGFR normale. Hanno quindi testato diverse generazioni di farmaci inibitori di EGFR. Solo l’afatinib, un inibitore di seconda generazione, ha spento in modo coerente l’attivazione di ERK1/2 e ridotto nettamente la crescita delle cellule tumorali. Nei modelli murini di tumori cerebrali e polmonari guidati da R252C, l’afatinib ha rallentato l’espansione tumorale e prolungato la sopravvivenza. In modo cruciale, quando il paziente originario — la cui malattia era peggiorata nonostante chemioterapia, un farmaco anti-vascolare e immunoterapia — è stato trattato con afatinib, le scansioni di polmone e cervello hanno mostrato una marcata riduzione del carico tumorale e il paziente ha beneficiato di diversi anni senza progressione.
Cosa significa per i pazienti
Questo lavoro rivela un modo finora non riconosciuto con cui una mutazione oncogena di EGFR può operare: graffando due recettori all’esterno della cellula e torcendoli in una posa attiva che accende direttamente ERK1/2 anziché seguire la catena di segnalazione canonica. Per i non specialisti, il punto chiave è che non tutte le mutazioni nello stesso gene si comportano allo stesso modo, e alcuni cambiamenti rari possono rispondere al meglio a farmaci mirati specifici. EGFR R252C sembra generare tumori particolarmente vulnerabili all’afatinib. Sebbene questa conclusione si basi attualmente su un caso paziente dettagliato e su ampi lavori di laboratorio, indica la strada verso test più mirati delle mutazioni del dominio esterno di EGFR e suggerisce che terapie target accuratamente selezionate potrebbero offrire nuova speranza a pazienti selezionati con tumori cerebrali e polmonari difficili da trattare.
Citazione: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Parole chiave: mutazione EGFR, glioma, cancro del polmone, segnalazione ERK, afatinib