Clear Sky Science · it
Migliorare la predizione con punteggi poligenici per gruppi sottorappresentati tramite trasferimento di apprendimento
Perché il tuo punteggio di rischio genetico potrebbe non funzionare per te
I «punteggi di rischio» genetici sono sempre più usati per stimare la probabilità che una persona sviluppi condizioni comuni come diabete, malattie cardiache o ipertensione. Ma la maggior parte di questi punteggi è stata costruita usando dati genetici di persone di ascendenza europea. Di conseguenza, spesso predicono male per persone di altre origini, sollevando preoccupazioni su equità e utilità nella pratica clinica. Questo studio pone una domanda semplice: possiamo riutilizzare ciò che abbiamo appreso da grandi dataset europei per costruire punteggi genetici migliori e più equi per i gruppi sottorappresentati—senza condividere i dati grezzi di nessuno?
Dai milioni di marcatori del DNA a un singolo punteggio di rischio
Un punteggio poligenico è come una pagella che somma i piccoli effetti di molti marcatori genetici distribuiti nel genoma. A ogni marcatore viene assegnato un peso che riflette quanto è associato a un tratto, sulla base di grandi studi genetici. Quando questi studi riguardano soprattutto europei, il punteggio risultante tende a funzionare meglio negli europei. Differenze negli sfondi genetici—quanto sono comuni certe varianti del DNA e come vengono ereditate insieme—significano che gli stessi pesi spesso non funzionano bene in popolazioni afroamericane, ispaniche e altre. Raccogliere dataset altrettanto ampi per ogni gruppo è costoso e lento, quindi gli autori hanno adottato una strategia di machine learning chiamata transfer learning: invece di ricominciare da zero per ogni popolazione, perfezionano un modello esistente addestrato altrove.
Come prendere in prestito conoscenza senza condividere i dati grezzi
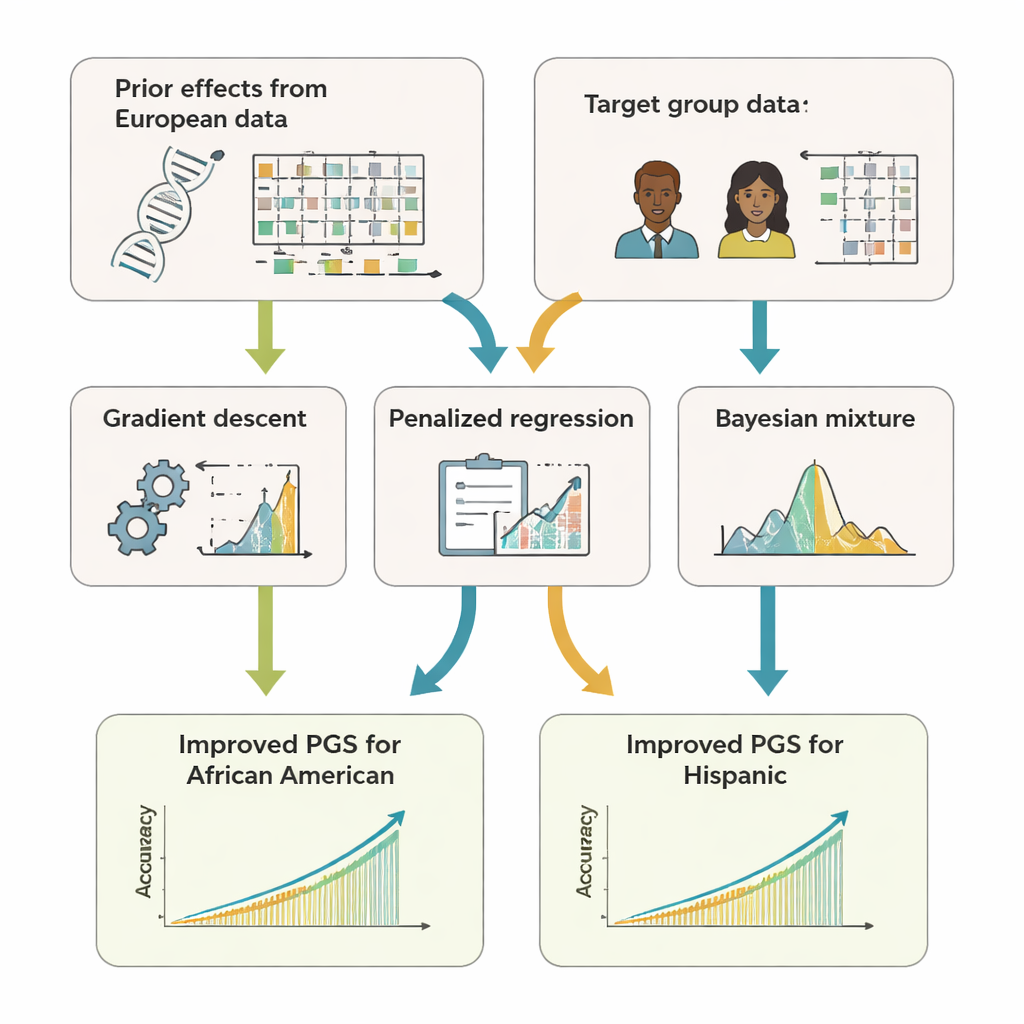
Il team ha sviluppato GPTL, un pacchetto software open-source in R che implementa tre approcci di transfer learning per i punteggi genetici. Tutti e tre partono da stime esistenti degli effetti del DNA ottenute in un ampio dataset di ascendenza europea e poi aggiustano delicatamente quelle stime usando dati di un gruppo target, come afroamericani o ispanici. Un metodo modifica i pesi europei passo passo usando la discesa del gradiente e si ferma precocemente, prima di sovrascriverli completamente. Un secondo metodo, chiamato regressione penalizzata, tira attivamente le nuove stime verso i valori originali a meno che i dati del gruppo target non forniscano prove solide del contrario. Il terzo, un modello di miscela bayesiano, permette a ogni marcatore del DNA di scegliere tra diverse fonti di informazione—come più gruppi di ascendenza o anche un’opzione «nessun effetto»—e le combina in base a quanto bene spiegano i dati del gruppo target.
Mettere i metodi alla prova
Per valutare l’efficacia di questi approcci, gli autori hanno usato sia simulazioni al computer sia dati reali di centinaia di migliaia di volontari nel UK Biobank e nel programma di ricerca statunitense All of Us. Si sono concentrati sui partecipanti afroamericani e ispanici come gruppi target e hanno usato i dati di ascendenza europea come principale fonte di informazione a priori. Su 11 tratti—compresi altezza, indice di massa corporea, lipidi nel sangue, pressione arteriosa e marcatori renali—i punteggi ottenuti con transfer learning hanno predetto in modo costantemente migliore rispetto ai punteggi costruiti solo all’interno del gruppo target o semplicemente riutilizzati dagli europei. Spesso, la loro accuratezza ha pareggiato o superato leggermente quella di metodi «multi-ascendenza» più complessi che richiedono la combinazione di dati grezzi da più popolazioni. Crucialmente, i metodi di GPTL richiedono solo statistiche sommarie—numeri aggregati sugli effetti genetici—quindi le istituzioni possono collaborare senza esporre dati genetici a livello individuale.
Quando più DNA non è sempre meglio
I ricercatori hanno anche esaminato come scegliere al meglio quali marcatori genetici includere. Contrariamente alla convinzione comune che usare ogni marcatore disponibile aiuti sempre, hanno scoperto che per i gruppi afroamericani e soprattutto ispanici l’inclusione di milioni di segnali molto deboli può effettivamente peggiorare le prestazioni, in particolare quando si usano rappresentazioni molto semplificate delle correlazioni genetiche. Concentrarsi su marcatori meglio supportati e usare informazioni più ricche su come le varianti sono ereditate insieme ha spesso prodotto punteggi più accurati. Lo studio ha inoltre mostrato che aggiungere informazioni a priori da più gruppi di ascendenza e modellare attentamente le differenze tra popolazioni migliora ulteriormente le predizioni.
Cosa significa questo per una predizione del rischio genetico più equa
Per le popolazioni non europee, i punteggi di rischio genetico pronti all’uso possono oggi prestarsi peggio di molto, ampliando potenzialmente le disparità di salute. Questo lavoro dimostra che il transfer learning—rifinire intelligentemente i punteggi basati su popolazioni europee usando dataset modesti dei gruppi sottorappresentati—può colmare gran parte di quel divario. In pratica, ciò significa che sistemi sanitari e ricercatori possono costruire strumenti genetici più accurati ed equi senza mettere in comune i dati grezzi tra istituzioni o ascendenze, riducendo le preoccupazioni sulla privacy. Sebbene nessun metodo sia ideale per ogni tratto e popolazione, il toolkit GPTL mostra che una predizione genetica più equa è tecnicamente alla portata se trattiamo i modelli passati non come prodotti fissi, ma come punti di partenza adattabili a tutti.
Citazione: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
Parole chiave: punteggi di rischio poligenico, transfer learning, predizione genetica, disparità di salute, genetica delle popolazioni