Clear Sky Science · it
Stima della stabilità del ripiegamento proteico con considerazione esplicita degli stati denaturati
Perché la stabilità delle proteine è importante
Ogni proteina nel tuo corpo è una piccola macchina molecolare che deve ripiegarsi in una precisa forma tridimensionale per funzionare correttamente. Se quel ripiegamento è troppo fragile, la proteina può funzionare male, aggregarsi o non essere prodotta affatto—problemi collegati a malattie e a insuccessi nella produzione di farmaci e enzimi a base proteica. Misurare quanto sia stabile una proteina in laboratorio è un processo lento e complesso, quindi gli scienziati cercano metodi informatici che possano indicare in modo affidabile, partendo solo dalla sequenza, quanto facilmente una proteina si disfi.
Un nuovo sguardo su proteine ripiegate e denaturate
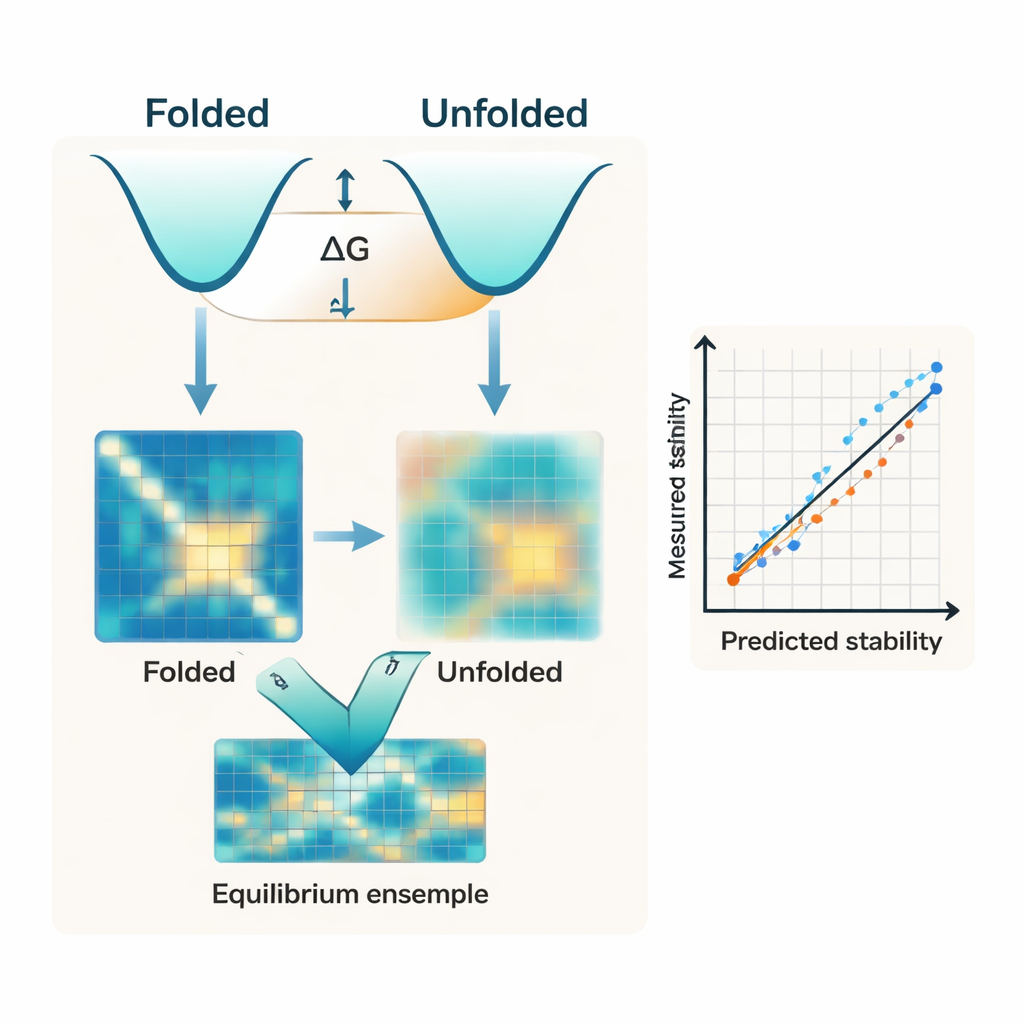
La maggior parte degli algoritmi moderni si concentra quasi esclusivamente sulla forma ripiegata di una proteina. Spesso partono da una struttura predetta con l’IA, come quelle di AlphaFold, e trattano quella singola struttura come il principale determinante della stabilità. Ma la stabilità è in realtà il divario energetico tra due ampie insiemi: lo stato compatto ripiegato e le molte forme flessibili che costituiscono lo stato denaturato. Gli autori sostengono che ignorare il lato denaturato di questo equilibrio sia una ragione chiave per cui gli strumenti esistenti faticano a corrispondere alle misure sperimentali dell’energia libera di ripiegamento, nota come ΔG.
Un nuovo modello che apprende entrambi gli stati
I ricercatori presentano IFUM, un sistema di deep learning progettato per stimare ΔG imparando al contempo quale sia l’equilibrio tra stati ripiegati e denaturati per ogni proteina. Invece di trattare lo stato denaturato come uno sfondo vago, IFUM usa concetti della fisica dei polimeri per rappresentarlo come una “coila casuale” e codifica entrambi gli stati, ripiegato e denaturato, come mappe di distanze tra coppie di amminoacidi. Il modello riceve informazioni da potenti reti pre‑addestrate su sequenze e strutture, quindi predice congiuntamente la stabilità totale e una mappa di probabilità che descrive quanto della popolazione proteica è ripiegata rispetto a denaturata per ogni coppia di residui. L’addestramento su un dataset molto ampio di piccole proteine caratterizzate sperimentalmente e di proteine note per essere disordinate aiuta IFUM a riconoscere sia le sequenze ben strutturate sia quelle flessibili.
Numeri più accurati e copertura più ampia delle mutazioni
Quando testato su un dataset accuratamente controllato di piccole proteine, IFUM predice i valori sperimentali di ΔG con errore minore e correlazione migliore rispetto ai precedenti metodi basati sull’IA che si affidano solo alla struttura ripiegata o a modelli linguistici addestrati sulle sequenze. In modo cruciale, il modello gestisce anche una grande varietà di cambiamenti di sequenza. Cattura con precisione gli effetti di mutazioni puntiformi singole e doppie, così come inserzioni e delezioni che modificano la lunghezza della proteina—situazioni in cui molti strumenti esistenti falliscono del tutto o non sono stati progettati per operare. Un confronto interno mostra che rimuovere l’obiettivo relativo allo stato denaturato peggiora significativamente le prestazioni, sottolineando che modellare esplicitamente l’insieme denaturato non è solo una scelta concettuale, ma centrale per l’accuratezza delle previsioni.
Dalla panca di progettazione ai test nel mondo reale
Per verificare se IFUM può guidare l’ingegneria proteica reale, gli autori lo applicano a tre difficili problemi di progettazione: stabilizzare le proteine interferone‑lambda, rimodellare la proteina di segnalazione immunitaria IL‑10 e migliorare un enzima modificatore di zuccheri chiamato UGT76G1. In tutti e tre i casi, le stabilità previste da IFUM seguono bene le temperature di fusione misurate, che indicano quanta temperatura una proteina può sopportare prima di denaturarsi. Il modello aiuta anche a selezionare centinaia di proteine completamente nuove progettate al computer per individuare quelle più probabilmente in grado di ripiegarsi e rimanere solubili nelle cellule, superando i punteggi di confidenza largamente usati provenienti dalle reti di predizione strutturale. Questi risultati suggeriscono che IFUM può essere usato come un pratico “filtro di stabilità” insieme ai controlli basati sulla struttura nei moderni flussi di lavoro di progettazione proteica.
Limiti e direzioni future
Come ogni modello, IFUM ha dei limiti. È addestrato principalmente su proteine corte, a catena singola e solubili, e i suoi valori assoluti di stabilità diventano meno affidabili per proteine molto più grandi o per quelle con ampi loop flessibili o regioni transmembrana. La sua descrizione dello stato denaturato rimane un modello statistico semplificato piuttosto che un quadro pienamente realistico di tutte le forme possibili. Tuttavia, l’approccio dimostra che insegnare a un’IA a considerare sia gli insiemi ripiegati sia quelli denaturati produce stime di stabilità più affidabili. Per i non esperti, la conclusione chiave è che IFUM ci avvicina alla possibilità di chiedere a un computer, con fiducia quantitativa, “Questo progetto proteico si manterrà realmente insieme?”, accelerando potenzialmente lo sviluppo di farmaci biologici più sicuri e di enzimi industriali più robusti.
Citazione: Lee, H., Cho, Y., Yun, J. et al. Protein folding stability estimation with explicit consideration of unfolded states. Nat Commun 17, 1883 (2026). https://doi.org/10.1038/s41467-026-68637-4
Parole chiave: stabilità delle proteine, ripiegamento delle proteine, apprendimento profondo, progettazione proteica, mutazioni