Clear Sky Science · it
TET1 come regolatore principale che controlla la sorveglianza della ferroptosi dipendente e indipendente da GPX4 nella leucemia mieloide acuta
Perché questa ricerca conta per il trattamento del cancro
Molti nuovi farmaci oncologici mirano a indurre nelle cellule maligne una modalità di autodistruzione chiamata ferroptosi, un tipo di morte cellulare guidata dal ferro e dal danneggiamento dei lipidi. Tuttavia alcuni tumori resistono a questo approccio. Questo studio rivela come una proteina che modifica il DNA chiamata TET1 aiuti le cellule leucemiche a eludere la ferroptosi tramite due distinti sistemi di difesa biochimici—e mostra che bloccare queste difese può rendere vulnerabili anche le neoplasie resistenti.
Un mix letale di ferro e lipidi ossidati
La ferroptosi si verifica quando il ferro alimenta l’ossidazione incontrollata dei lipidi nelle membrane cellulari, provocando infine la lisi delle cellule. Nella leucemia mieloide acuta (LMA), come in molti tumori, le cellule dispongono di potenti sistemi di sorveglianza per tenere sotto controllo questo processo. Uno dei guardiani principali è l’enzima GPX4, che utilizza una piccola molecola chiamata glutatione per neutralizzare i perossidi lipidici dannosi. Altri sistemi di riserva generano molecole antiossidanti in grado di intrappolare radicali pericolosi anche quando GPX4 è compromesso. Capire quali interruttori principali coordinano queste difese è cruciale per progettare terapie che inducano in modo affidabile la ferroptosi nelle cellule tumorali preservando i tessuti sani.
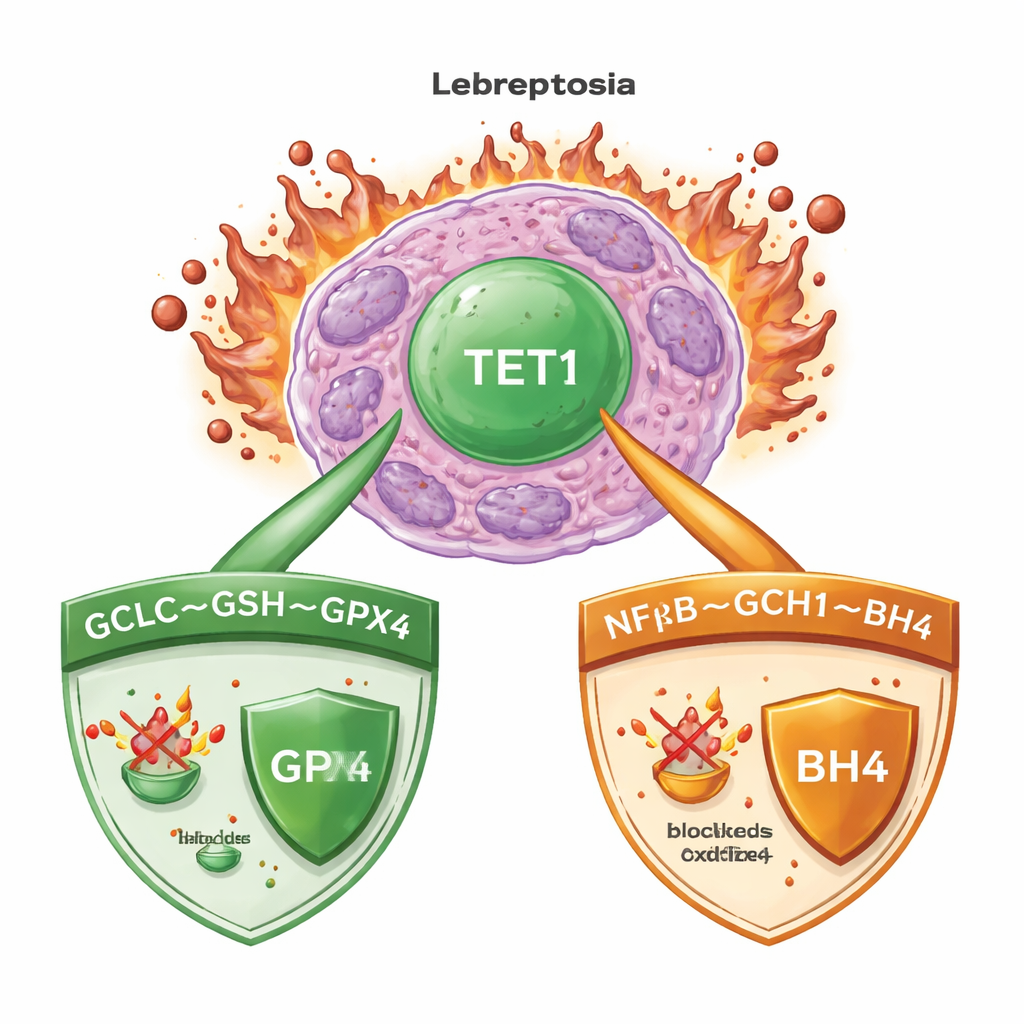
TET1 emerge come hub di controllo centrale
I ricercatori hanno confrontato dozzine di campioni tumorali, incluse molte linee di LMA e cellule derivate da pazienti, e hanno osservato un chiaro schema: le cellule che resistevano alla ferroptosi avevano livelli più alti di TET1, un enzima che modifica marchi chimici sul DNA e influenza l’attività genica. Quando hanno ridotto i livelli di TET1 con strumenti genetici o ne hanno inibito l’attività con una piccola molecola, le cellule tumorali sono diventate sensibilmente più suscettibili ai farmaci che inducono la ferroptosi. Questo effetto è stato osservato sia in colture cellulari sia in modelli murini di LMA. Al contrario, aumentare l’espressione di TET1 ha protetto le cellule dalla morte ferroptotica e ha limitato l’accumulo di specie reattive dell’ossigeno, i sottoprodotti chimicamente aggressivi che guidano il danno alle membrane.
Rafforzare lo scudo antiossidante principale
Approfondendo, il team ha mappato dove agisce TET1 sul genoma e ha scoperto che attiva direttamente un gene chiamato GCLC. GCLC codifica un enzima critico che avvia la produzione di glutatione, il combustibile per GPX4. Aumentando un marchio specifico sul DNA (5-idrossimetilcitosina) nel promotore di GCLC, TET1 potenzia la sintesi del glutatione. In condizioni normali di nutrienti questo aumenta la principale riserva antiossidante della cellula. In condizioni di deprivazione di cistina, lo stesso complesso enzimatico produce anche γ-glutamil–peptidi insoliti che aiutano a eliminare l’eccesso di glutammato, un altro modo per attenuare la ferroptosi. Sia nelle cellule in coltura sia nei topi, la perdita di TET1 o l’inibizione farmacologica della sintesi del glutatione ha ridotto drasticamente i livelli di glutatione e questi peptidi protettivi, rendendo le cellule leucemiche molto più vulnerabili agli induttori di ferroptosi.
Una seconda via di fuga indipendente da GPX4
Con sorpresa, il ruolo protettivo di TET1 non si limitava all’asse glutatione–GPX4. Anche quando GPX4 veniva rimosso dalle cellule leucemiche, livelli elevati di TET1 riuscivano ancora a prevenire la morte ferroptotica, suggerendo una seconda linea di difesa. Gli autori hanno ricondotto questo effetto all’attivazione da parte di TET1 della via di segnalazione NFκB, in particolare di una componente chiamata NFKB2. Quest’ultima a sua volta aumenta l’espressione di GCH1, un enzima che produce la molecola antiossidante BH4. BH4 può proteggere i lipidi di membrana dall’ossidazione senza dipendere da GPX4. Quando GCH1 è stato silenziato geneticamente o bloccato chimicamente, la capacità di TET1 di proteggere le cellule dalla ferroptosi è risultata parzialmente compromessa. Complessivamente, questi risultati definiscono una via TET1–NFKB2–GCH1 che costituisce un sistema di sorveglianza della ferroptosi indipendente da GPX4.
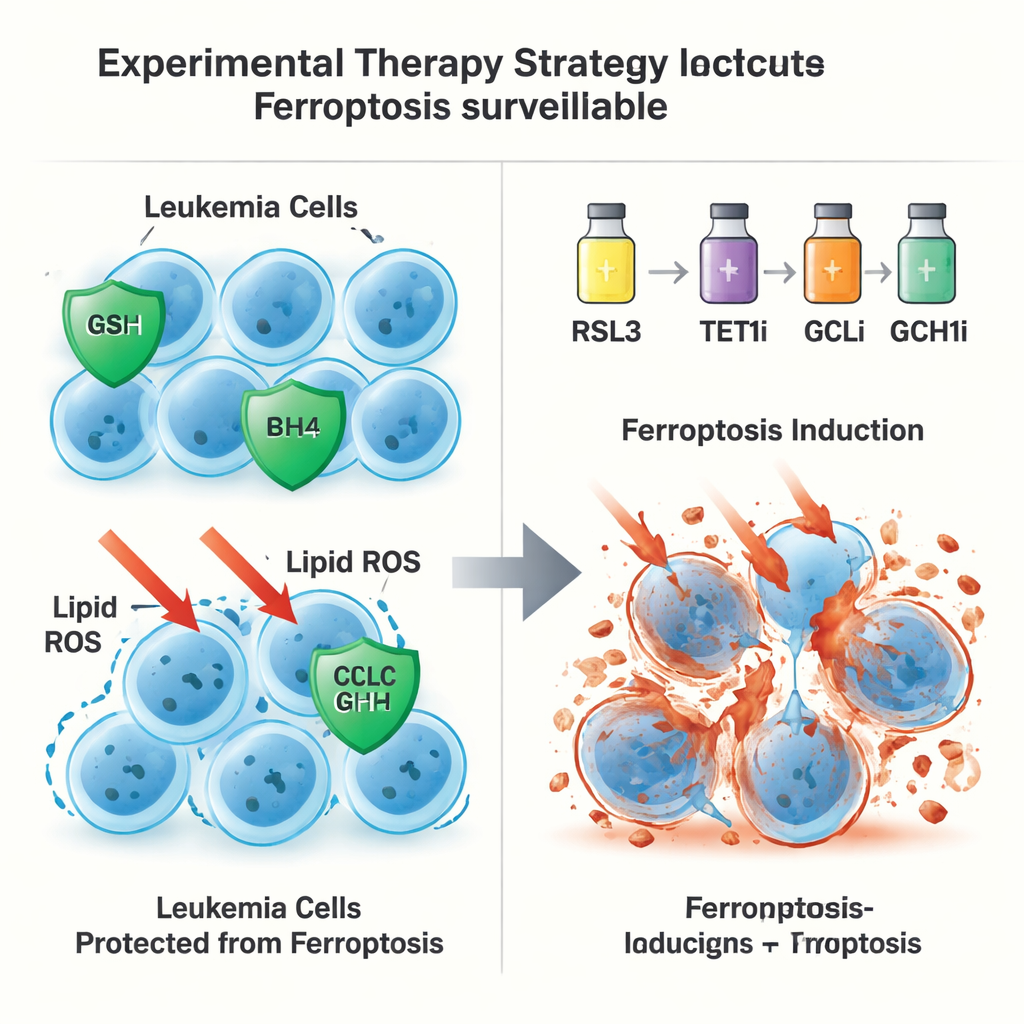
Trasformare una debolezza in un’opportunità terapeutica
Con questa mappatura a doppio percorso, i ricercatori hanno testato se stimolare simultaneamente la ferroptosi e indebolire le difese controllate da TET1 potesse offrire un vantaggio terapeutico. Nei modelli murini di LMA e in innesti leucemici derivati da pazienti, basse dosi di un farmaco che induce la ferroptosi combinate con inibitori di TET1, della sintesi del GSH (tramite GCLC) o di GCH1 hanno ridotto drasticamente il carico di leucemia, prolungato la sopravvivenza e depauperato le popolazioni di cellule iniziatrici leucemiche. È importante notare che l’induttore di ferroptosi è stato usato a una frazione delle dosi riportate in studi precedenti, riducendo le preoccupazioni sulla tossicità per le cellule staminali ematiche normali.
Cosa significa per le future terapie oncologiche
Per i non specialisti, il messaggio chiave è che le cellule leucemiche sopravvivono attivando due sistemi antiossidanti sovrapposti, entrambi coordinati da TET1: uno centrato su glutatione e GPX4 e l’altro basato su GCH1 e BH4. Questo lavoro mostra che modulando moderatamente la ferroptosi e bloccando simultaneamente TET1 e i suoi partner a valle, i medici potrebbero un giorno superare la resistenza e spingere selettivamente le cellule tumorali al punto di non ritorno, senza sovraccaricare i tessuti sani. Sebbene queste strategie non siano ancora pronte per la clinica, lo studio identifica TET1 come un potente nodo di controllo e un promettente bersaglio per terapie combinatorie nella LMA e possibilmente in altri tumori difficili da trattare.
Citazione: Yang, L., Lu, J., Yun, W. et al. TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia. Nat Commun 17, 1800 (2026). https://doi.org/10.1038/s41467-026-68509-x
Parole chiave: ferroptosi, leucemia mieloide acuta, TET1, glutatione, epigenetica del cancro