I tumori polmonari resistenti agli inibitori di EGFR mostrano sensibilità collaterale a un ponte molecolare covalente e indipendente dalla cisteina che oligomerizza KEAP1
Perché il cancro polmonare resistente ai farmaci è importante
I farmaci mirati hanno trasformato il trattamento di alcuni tumori polmonari concentrandosi su un segnale di crescita difettoso chiamato EGFR. Tuttavia, per la maggior parte dei pazienti questi farmaci smettono di funzionare entro pochi anni perché il tumore evolve resistenza. Questo studio scopre una svolta sorprendente: una volta che i tumori diventano resistenti agli inibitori di EGFR, sviluppano un nuovo tallone d’Achille che può essere attaccato con un tipo diverso di composto. Comprendere questa debolezza nascosta potrebbe ispirare strategie terapeutiche future che intrappolino l’evoluzione del cancro invece di inseguirla continuamente.
Una debolezza nascosta rivelata
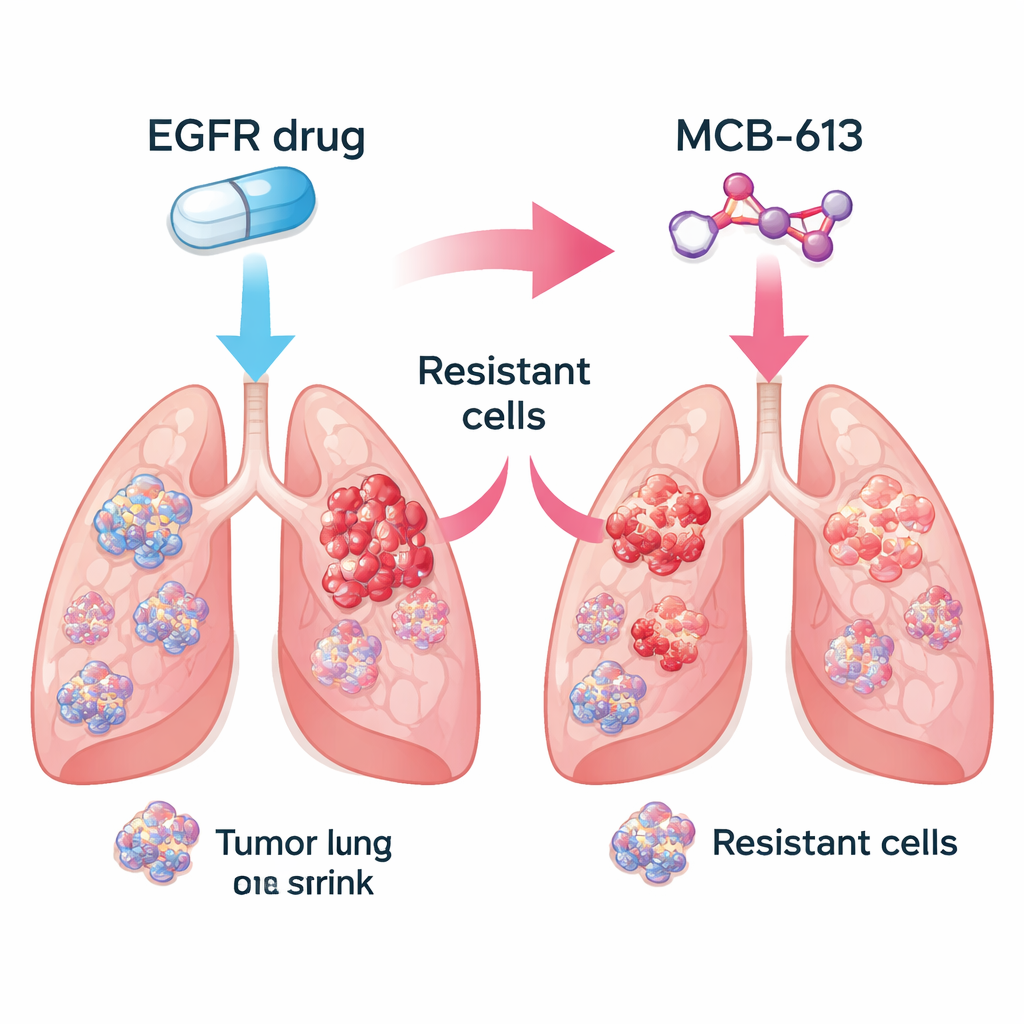
I ricercatori si sono concentrati sui carcinomi polmonari non a piccole cellule guidati da EGFR mutato, una forma comune della malattia. In laboratorio hanno confrontato cellule tumorali sensibili ai farmaci con cellule correlate che avevano evoluto resistenza a farmaci che bloccano EGFR come gefitinib e osimertinib. Hanno poi testato una libreria di circa 2.100 piccole molecole per vedere quali uccidevano le cellule resistenti più efficacemente rispetto alle cellule sensibili originali. Tra molti candidati, un composto chiamato MCB-613 è emerso costantemente. Le cellule resistenti che ignoravano gli inibitori di EGFR si sono rivelate insolitamente vulnerabili a MCB-613, sia in piastre da laboratorio sia in tumori murini.
Intrappolare popolazioni tumorali miste Figure 1.
I tumori reali sono miscele di cellule: alcune restano sensibili al farmaco originale, mentre altre acquisiscono resistenza tramite diversi stratagemmi genetici. Il gruppo si è chiesto se combinare un inibitore di EGFR con MCB-613 potesse eliminare questa diversità. In un esperimento controllato hanno miscelato per lo più cellule sensibili con una piccola frazione di più tipi resistenti, imitando il tumore di un paziente. Trattare questa popolazione mista con l’inibitore di EGFR o con MCB-613 da soli ha permesso ad alcune cellule di sopravvivere e proliferare. Ma quando i due agenti sono stati usati insieme, l’intera popolazione è collassata. Questo suggerisce che abbinare una terapia mirata standard a un farmaco di “sensibilità collaterale” scelto con cura potrebbe spingere i tumori in un vicolo cieco evolutivo.
Un ponte molecolare che rompe un guardiano
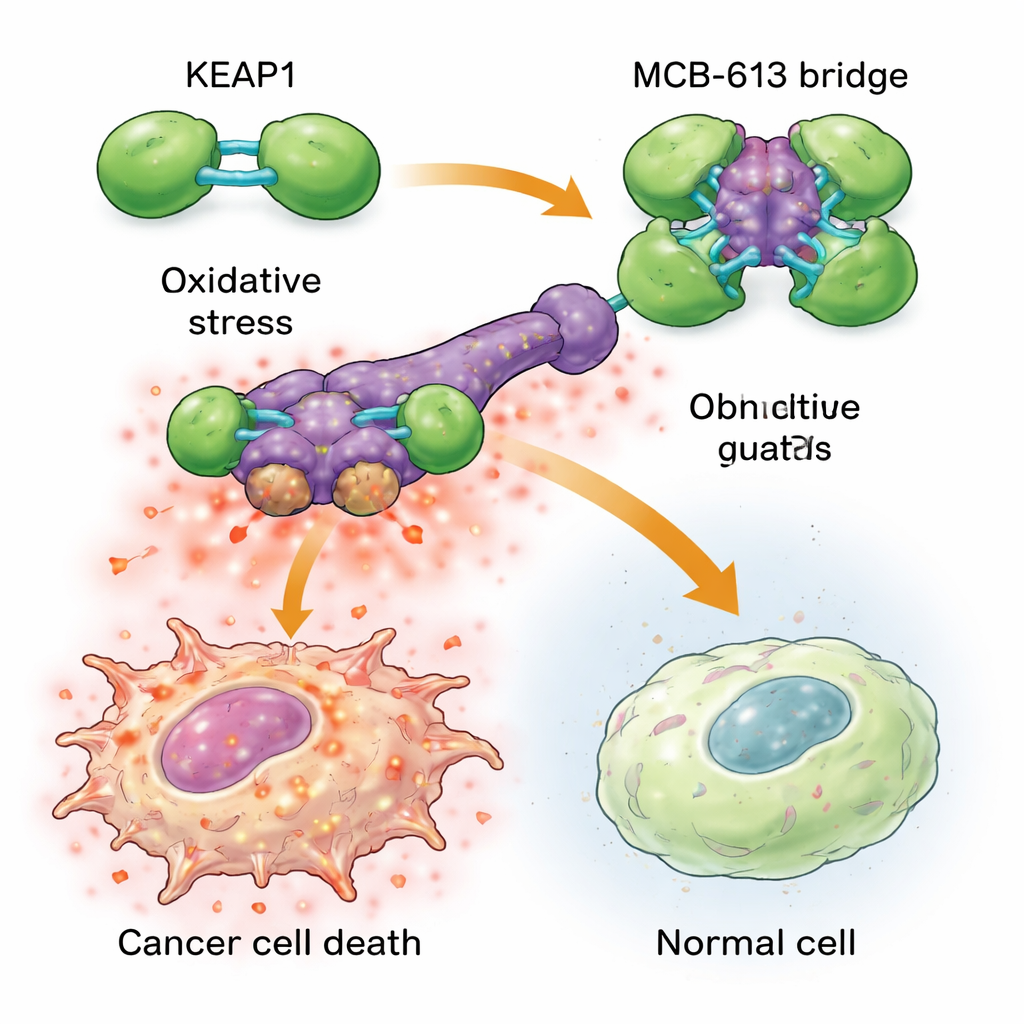
Per capire perché MCB-613 colpisce così duramente le cellule resistenti, gli scienziati hanno esaminato a quali proteine si lega. Usando sonde chimiche e uno screen mirato CRISPR, hanno individuato una proteina chiamata KEAP1 come essenziale per l’effetto di MCB-613. KEAP1 normalmente funziona da guardiano cellulare, percependo lo stress e aiutando a regolare risposte protettive. Il team ha scoperto che MCB-613 si lega a KEAP1 in modo insolito: si comporta come un ponte molecolare rigido che collega le unità di KEAP1 formando aggregati sovradimensionati e anomali. Questo processo non dipende dai classici siti reattivi contenenti zolfo in KEAP1, ma da una specifica lisina nella regione di dimerizzazione. Quando quella lisina è stata mutata, MCB-613 non è più riuscito a far aggregare KEAP1 e le cellule resistenti non erano più ipersensibili al composto.
Trasformare uno stress utile in un sovraccarico letale Figure 2.
L’aggregazione di KEAP1 innesca una catena di eventi pericolosa nelle cellule tumorali resistenti ai farmaci. Queste cellule vivono già sotto un livello di stress di base più elevato, con livelli aumentati di specie reattive dell’ossigeno (sottoprodotti chimici dannosi) e attività incrementata di una rete protettiva nota come integrated stress response. Quando si aggiunge MCB-613, la perturbazione di KEAP1 fa precipitare questo stato stressante oltre il limite: le specie reattive dell’ossigeno si accumulano ulteriormente e regolatori chiave dello stress come ATF4 e CHOP attivano programmi mortali potenti. Bloccare questi regolatori dello stress, o neutralizzare chimicamente le specie reattive dell’ossigeno, ha protetto in gran parte le cellule da MCB-613. È interessante che il partner classico di KEAP1, NRF2, spesso considerato il motore principale delle difese antiossidanti, non fosse responsabile della morte; in realtà, rimuovere NRF2 rese le cellule ancora più sensibili, sottolineando che MCB-613 sfrutta una via diversa e non canonica.
Cosa potrebbe significare per i trattamenti futuri
Per ora, MCB-613 è un composto di laboratorio con difetti chimici che lo rendono inadatto come farmaco. Ma rivela un concetto potente: man mano che i tumori polmonari evolvono resistenza agli inibitori di EGFR, possono restare intrappolati in uno stato di stress che può essere selettivamente preso di mira da composti che forzano KEAP1 in assemblaggi disfunzionali. In linea di principio, versioni perfezionate di tali “ponti molecolari” potrebbero essere sviluppate per essere più sicure e precise, offrendo agli oncologi un modo per guidare i tumori verso un’“impossibile scelta” tra sensibilità alla terapia mirata originale e sensibilità all’agente successivo che induce stress. Questa strategia di intrappolamento evolutivo potrebbe infine aiutare a ritardare o superare la resistenza nel cancro polmonare con mutazione EGFR e forse in altri tumori difficili da trattare.
Citazione: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1
Parole chiave: cancro polmonare con mutazione EGFR, resistenza ai farmaci, sensibilità collaterale, KEAP1, stress ossidativo