Clear Sky Science · it
Usare i riferimenti lineari del pangenoma per scoprire varianti mancanti dell’autismo
Perché i cambiamenti nascosti del DNA sono importanti per l’autismo
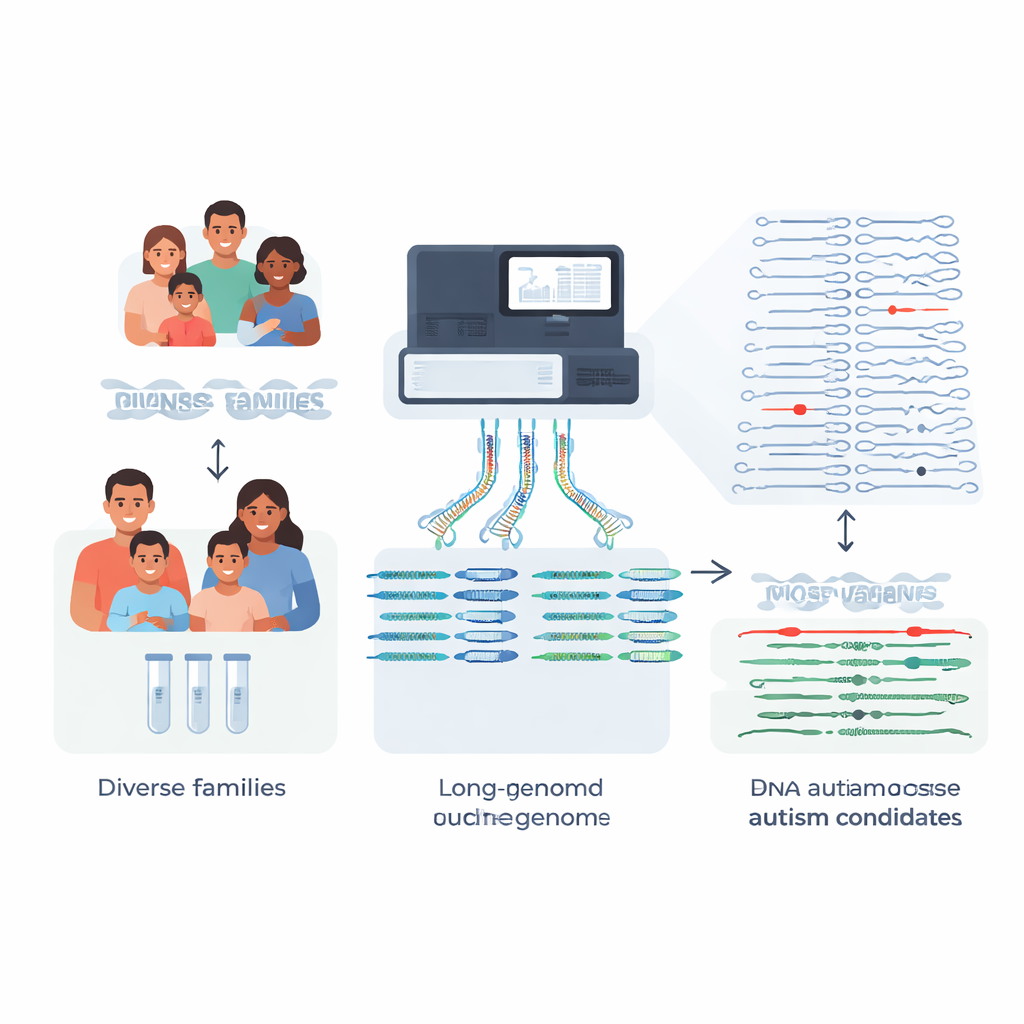
La maggior parte delle famiglie che si sottopone a test genetici per un bambino autistico spera di ottenere risposte chiare, ma circa quattro su cinque non ricevono una spiegazione genetica definitiva. Questo studio affronta una causa chiave: molte variazioni del DNA con impatto rilevante sono troppo complesse per essere rilevate dai test standard. Ricostruendo genomi quasi completi per 189 persone di 51 famiglie con casi di autismo e confrontandoli con un nuovo e più ricco riferimento “pangenoma”, i ricercatori mostrano come il sequenziamento avanzato possa svelare mutazioni rare e precedentemente invisibili che potrebbero contribuire a spiegare alcuni casi di autismo e condizioni correlate.
Oltre i test genetici standard
I test clinici tradizionali si basano su brevi frammenti di DNA per scandagliare il genoma di una persona. Questo approccio funziona bene per molte variazioni a singola lettera, ma spesso fallisce nelle regioni ripetitive o strutturalmente complesse, proprio dove si nascondono alcune mutazioni fortemente patogenetiche. Il gruppo si è concentrato su famiglie in cui precedenti test basati su short-read del genoma, dell’esoma o su pannelli genici non avevano individuato una causa per sintomi di autismo o simili alla sindrome di Rett. Utilizzando il sequenziamento a letture lunghe, che legge tratti molto più estesi di DNA, hanno costruito assemblaggi genomici fagici e di alta qualità per 189 individui. Ciò ha permesso di ricostruire le due copie di ciascun cromosoma per ogni persona, una ereditata da ciascun genitore, con pochissime lacune.
Varianti strutturali: cambiamenti grandi con grandi effetti
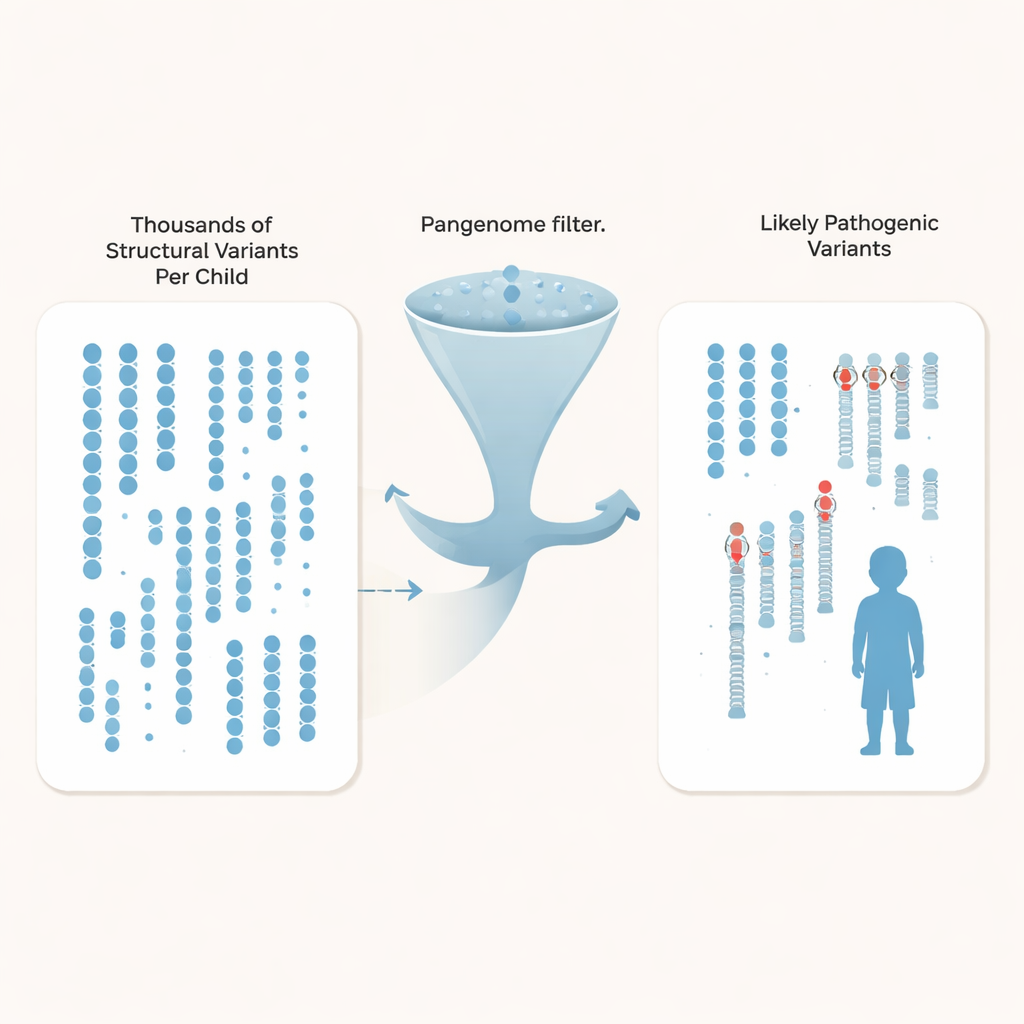
Invece di limitarsi alle differenze a singola lettera, i ricercatori si sono concentrati sulle varianti strutturali—inserzioni, delezioni e riorganizzazioni che coinvolgono almeno 50 basi di DNA e possono disturbare i geni o i loro elementi regolatori. Ogni bambino presentava circa 27.000 di queste varianti, ma la grande maggioranza sono differenze innocue condivise nella popolazione. Confrontando le loro famiglie affette con centinaia di genomi di controllo del pangenoma sequenziati in profondità e provenienti da ancestrie diverse, il team è stato in grado di filtrare oltre il 97% delle varianti strutturali comuni per ciascun bambino, lasciando circa 600 candidati rari per genoma, e fino a circa 200 quando si utilizza il più ampio set di controllo.
Scoprire mutazioni perse in geni di rischio noti
Con lo spazio di ricerca drasticamente ridotto, gli autori hanno integrato diverse linee di evidenza: geni noti per l’autismo e i disturbi del neurosviluppo, regioni regolatorie attive nella corteccia umana in via di sviluppo e i modelli di ereditarietà all’interno di ogni famiglia. Hanno identificato tre mutazioni chiaramente patogenetiche che i test precedenti avevano mancato. Tra queste c’è un nuovo segnale di stop nel gene SYNGAP1, importante per la funzione sinaptica, e una delezione che elimina l’ultimo esone di MECP2, un gene chiave nella sindrome di Rett, nonostante il paziente avesse già sostenuto molteplici test clinici. Hanno inoltre confermato una variante causa-malattia in TBL1XR1, un gene che interagisce con MECP2. In totale, hanno evidenziato altre nove varianti strutturali—spesso ereditate e localizzate in regioni regolatorie vicino a geni legati al cervello—come forti candidate per futuri test funzionali.
Cosa lo studio non ha trovato—e perché ciò è comunque importante
Nonostante questa ricerca approfondita, gli autori non hanno osservato un chiaro eccesso complessivo di varianti strutturali nei bambini autistici rispetto ai loro fratelli non affetti, almeno con questa dimensione del campione modesta. C’era però un indizio di un maggior numero di cambiamenti strutturali sul cromosoma X nelle ragazze affette, e gli assemblaggi quasi completi di X e Y hanno permesso di individuare pattern insoliti come un’estrema deviazione dell’inattivazione del cromosoma X. Queste caratteristiche potrebbero diventare indizi importanti man mano che verranno studiate più famiglie. Fondamentalmente, il lavoro dimostra che il sequenziamento a letture lunghe può recuperare varianti patogenetiche che i metodi a letture corte perdono, specialmente nelle parti difficili del genoma e nelle regioni di controllo che modulano l’attività genica.
Cosa significa per le famiglie e per i futuri test
Per le famiglie, l’impatto immediato è modesto ma significativo: nei casi difficili da risolvere di questo studio, circa il 6% ha ricevuto una diagnosi genetica chiara e quasi una su cinque ha ottenuto nuovi forti candidati da investigare. Per il campo, il messaggio è più ampio. Con l’aggiunta di riferimenti genomici completi e più diversi al pangenoma e con la crescente accessibilità del sequenziamento a letture lunghe, i clinici potranno escludere rapidamente le variazioni strutturali comuni e concentrarsi su un piccolo insieme di varianti rare e potenzialmente dannose per ciascun paziente. Questo cambiamento potrebbe progressivamente trasformare molti degli attuali casi di autismo “irrisolti” in situazioni in cui la biologia sottostante—e le possibili vie di supporto e trattamento—sono molto meglio comprese.
Citazione: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Parole chiave: genetica dell’autismo, sequenziamento a letture lunghe, varianti strutturali, pangenoma umano, sindrome di Rett