Clear Sky Science · it
Prove interspecie per un’origine nello sviluppo dell’ipersonnia adulta con perdita delle molecole di adesione sinaptica beat-Ia/CADM2
Perché dormire troppo può essere un problema serio
Molti invidiano chi dorme molto, ma per chi soffre di ipersonnia idiopatica un sonno travolgente può rovinare lavoro, scuola e relazioni. Questa condizione lascia le persone esauste nonostante una notte completa di riposo, e i medici non ne comprendono ancora pienamente le cause. Questo studio combina genetica umana con esperimenti su moscerini della frutta e zebrafish per ricondurre la sonnolenza eccessiva al modo in cui i circuiti cerebrali si connettono durante lo sviluppo, indicando una possibile nuova strategia terapeutica.
Individuare geni legati alla sonnolenza nel DNA umano
I ricercatori hanno iniziato analizzando enormi studi genetici su centinaia di migliaia di persone che riportavano eccessiva sonnolenza diurna o sonnellini frequenti. Invece di assumere che il gene più vicino a ciascuna variante di rischio fosse rilevante, hanno esaminato quartieri genomici tridimensionali più ampi, detti “domini topologici”, per raccogliere tutti i geni plausibili. Hanno quindi utilizzato strumenti computazionali per trovare geni corrispondenti nei moscerini, ottenendo oltre 200 geni da testare. Silenziando sistematicamente questi geni in tutti i neuroni, hanno cercato moscerini che dormissero molto più del normale. Tra i risultati più forti sono emersi geni della famiglia “beaten path”, controparte del gene umano CADM2, che codifica per una molecola che aiuta le cellule nervose a aderire e connettersi alle sinapsi. 
Moscerini assonnati e pesci assonnati
Quando la versione del gene CADM2 nel moscerino, chiamata beat-Ia, veniva ridotta nei neuroni, i moscerini dormivano molto più a lungo sia di giorno che di notte. Non erano letargici durante la veglia; invece, i loro episodi di sonno erano più lunghi, era più difficile svegliarli e si riaddormentavano più rapidamente dopo l’accensione delle luci—caratteristiche che rispecchiano da vicino l’ipersonnia umana. Il team ha poi testato CADM2 negli zebrafish, un piccolo vertebrato il cui sonno può essere monitorato tramite video. La disruzione del gene fish cadm2b ha aumentato il sonno senza ridurre il movimento durante la veglia, supportando un ruolo conservato di questa molecola nel mantenere lo stato di attenzione degli animali.
Come il cablaggio cerebrale precoce forma il sonno per tutta la vita
Un’intuizione chiave è stata che beat-Ia è importante soprattutto durante lo sviluppo cerebrale, non nell’età adulta. Attivando il silenziamento del gene solo prima o solo dopo l’emergenza dei moscerini dallo stadio pupale, i ricercatori hanno mostrato che perturbare beat-Ia precocemente nella vita era sufficiente a causare ipersonnia lifelong, mentre ridurlo solo negli adulti aveva poco effetto. Hanno ricondotto l’azione di beat-Ia a un piccolo gruppo di neuroni che producono il neuropeptide F (NPF), la controparte del moscerino del neuropeptide Y (NPY) dei vertebrati. Nei moscerini normali, i neuroni NPF inviano densissime proiezioni sinaptiche in una regione cerebrale chiamata zona sottoesofagea, dove si connettono a specifici neuroni inibitori (produttori di GABA) che aiutano a stabilizzare la veglia. Nei moscerini privi di beat-Ia, i grandi aggregati di sinapsi in questa regione non si formavano, anche se le fibre nervose fisicamente raggiungevano l’area. Ciò suggerisce che una formazione sinaptica difettosa, più che un cablaggio macroscopicamente errato, possa spostare l’equilibrio verso un sonno eccessivo.
Da circuiti mal connessi a un bersaglio farmacologico
Usando una mappa dettagliata delle connessioni del cervello del moscerino, il team ha identificato una manciata di neuroni downstream nella zona sottoesofagea che ricevono input dalle cellule NPF e sono predetti essere GABAergici. Silenziare queste cellule aumentava il sonno, mentre attivarle promuoveva la veglia, coerente con l’idea che NPF normalmente mantenga la veglia attivando una rete inibitoria locale. I ricercatori si sono poi chiesti se fosse possibile compensare la perdita di funzione simile a CADM2 potenziando la segnalazione di NPY. Negli zebrafish carenti di cadm2b, il bagno dei larve in un farmaco che attiva un sottotipo di recettore NPY (simile al recettore NPF del moscerino) ha riportato il loro sonno eccessivo a livelli normali, senza influenzare fortemente i pesci normali. Questo risultato interspecie suggerisce che quando le molecole di adesione sinaptica vanno incontro a anomalie durante lo sviluppo, potenziare le vie NPY in età adulta potrebbe aiutare a ristabilire il giusto equilibrio sonno–veglia. 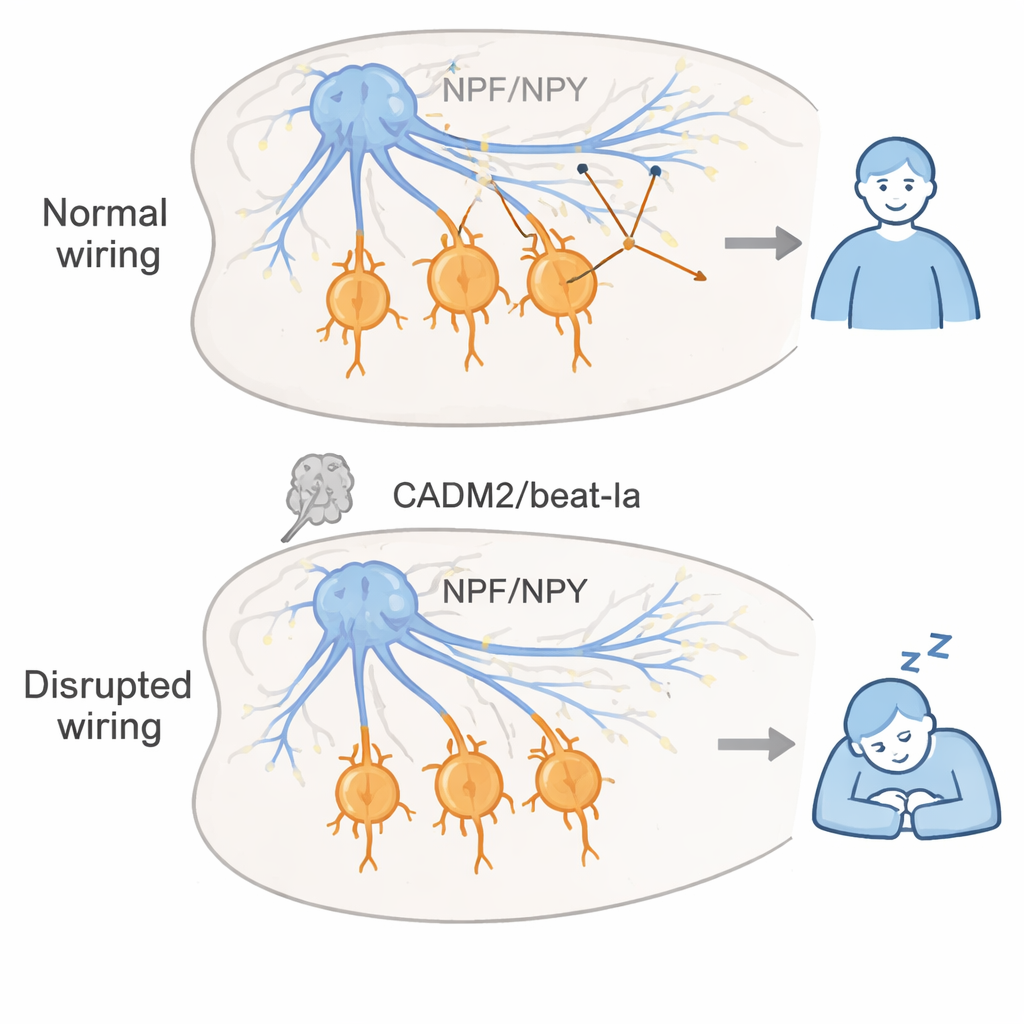
Cosa significa per le persone che non riescono a restare sveglie
Nel complesso, il lavoro propone che alcune forme di ipersonnia idiopatica possano derivare da errori sottili nel modo in cui i circuiti cerebrali che promuovono la veglia vengono cablati nella prima parte della vita, coinvolgendo CADM2 e molecole di adesione correlate. Questi cambiamenti non distruggono il cervello ma riconfigurano l’intensità delle comunicazioni tra determinate vie del sonno e dell’eccitazione. È importante che lo studio mostri anche che, sebbene il problema di cablaggio inizi nello sviluppo, i suoi effetti potrebbero comunque essere trattabili in seguito intervenendo su sistemi di neuropeptidi conservati come NPY. Per i pazienti, questo apre la possibilità che farmaci futuri progettati per modulare finemente questi percorsi di segnalazione possano offrire un sollievo più mirato dalla debilitante sonnolenza diurna.
Citazione: Mace, K., Zimmerman, A., Chesi, A. et al. Cross-species evidence for a developmental origin of adult hypersomnia with loss of synaptic adhesion molecules beat-Ia/CADM2. Nat Commun 17, 1628 (2026). https://doi.org/10.1038/s41467-026-68343-1
Parole chiave: ipersonnia idiopatica, genetica del sonno, adesione sinaptica, neuropeptide Y, sviluppo cerebrale