Clear Sky Science · it
Base molecolare dell’antagonismo del recettore umano dell’arginina vasopressina 1A in forma dimerica
Perché conta un recettore ormonale cerebrale
Ormoni come la vasopressina e l’ossitocina sono noti soprattutto per il controllo dell’equilibrio idrico, della pressione sanguigna, del parto e dei legami sociali. Tuttavia, il modo in cui funzionano i loro recettori a livello atomico è rimasto in gran parte nascosto. Questo articolo svela strutture 3D dettagliate di un recettore chiave, il recettore umano della vasopressina V1a, collegato al comportamento sociale, allo stress e a diversi disturbi cerebrali. Capirne la forma e il modo in cui i farmaci lo bloccano potrebbe aiutare gli scienziati a progettare terapie migliori per condizioni come l’autismo, il disturbo da stress post-traumatico e la malattia di Huntington.
Un recettore gemello che modula segnali di cuore, rene e cervello
Il recettore V1a si trova sulla superficie di molte cellule dell’organismo, in particolare nei vasi sanguigni, nei reni e in alcune regioni cerebrali. Quando l’ormone vasopressina vi si lega, il recettore attiva vie di segnalazione interne che controllano la pressione sanguigna, l’equilibrio dei fluidi e i circuiti cerebrali per l’interazione sociale, le emozioni e lo stress. Studi genetici e clinici hanno collegato un segnale V1a anomalo a disturbi dello spettro autistico, al PTSD e alla malattia di Huntington, rendendolo un bersaglio farmacologico interessante. Diversi farmaci antagonisti del V1a sono già in uso o in sperimentazione clinica, ma fino ad ora nessuno aveva osservato il recettore umano ad alta risoluzione, lasciando importanti domande su come si assembla e su come esattamente questi farmaci lo inattivano. 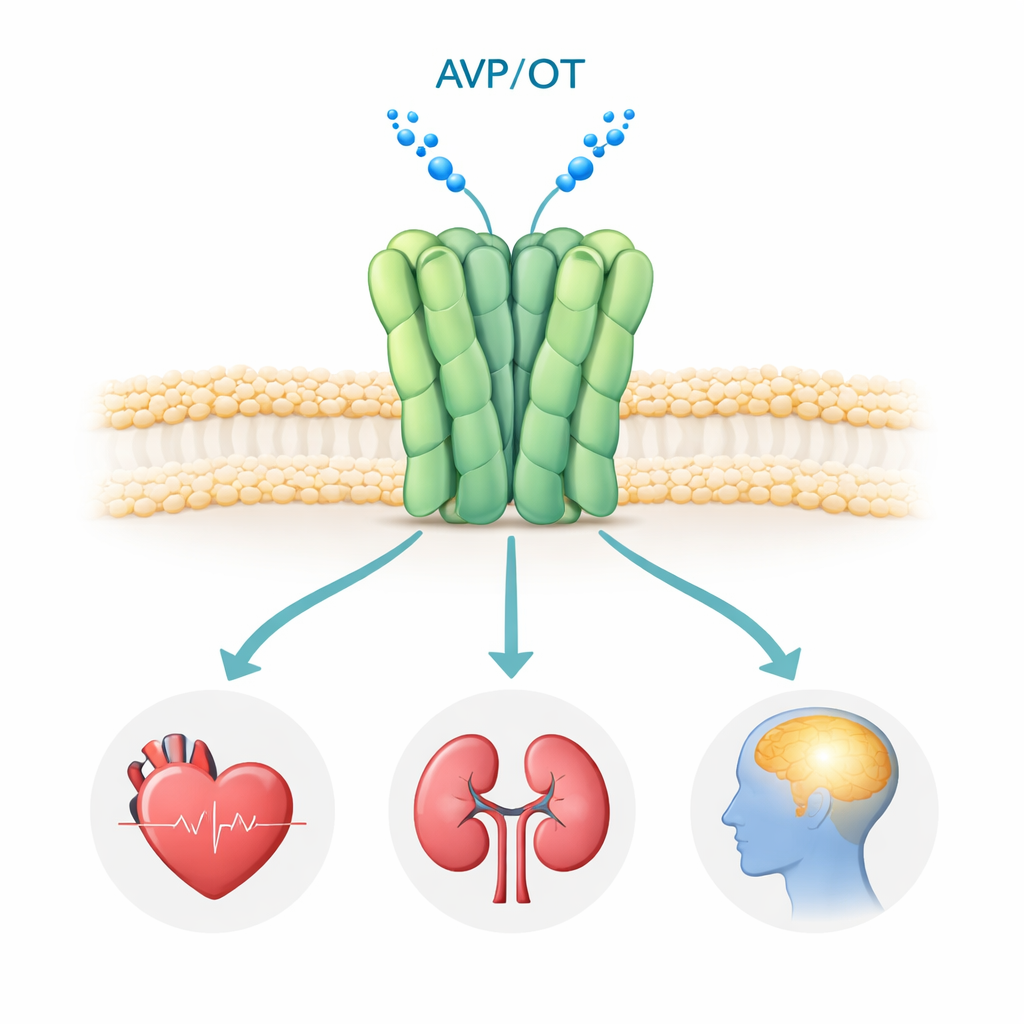
Cogliere la struttura del recettore in più stati legati ai farmaci
I ricercatori hanno usato la criomicroscopia elettronica, una tecnica che congela istantaneamente le proteine e le osserva con un fascio di elettroni, per visualizzare il recettore umano V1a. Per stabilizzare la proteina, hanno ingegnerizzato una forma leggermente modificata che mantiene una buona capacità di legare i farmaci e l’hanno accoppiata a un piccolo frammento anticorpale (un nanobody) per facilitare l’imaging. Hanno risolto strutture del recettore da solo e legato a tre antagonisti di rilievo medico: atosiban (un farmaco peptidico usato per prevenire il parto prematuro) e due piccole molecole in grado di attraversare la barriera ematoencefalica, balovaptan e SRX246, testate in persone con autismo o malattia di Huntington. Tutte le strutture hanno raggiunto una risoluzione prossima all’atomica, rivelando le posizioni delle sette eliche transmembrana del recettore, delle anse flessibili e dei farmaci legati.
Un recettore che preferisce agire in coppia
A differenza di recettori correlati visti finora solo come unità singole, il V1a è apparso come una coppia — un dimero — in tutte e quattro le strutture cryo-EM. I due recettori giacciono affiancati nella membrana, stabilendo contatti stretti principalmente attraverso una delle loro eliche, sostenuti da interazioni sia polari sia idrofobiche. Per verificare se questo accoppiamento avvenga anche nelle cellule vive, il team ha fuso V1a a una proteina fluorescente brillante e ha usato un metodo di fotobleaching a singola molecola: se un punto sulla superficie cellulare conteneva due copie del recettore, la sua luce sarebbe scomparsa in due passi. Circa tre quarti dei punti osservati si sono scoloriti esattamente in due passi, a forte sostegno dell’idea che V1a formi naturalmente dimeri sulla superficie cellulare. Quando gli scienziati hanno mutato residui chiave di contatto per perturbare l’interfaccia, il recettore è passato verso unità singole ed è diventato meno reattivo a ormone e farmaco, suggerendo che il dimero non è solo un ornamento strutturale ma ha importanza funzionale. 
Un cancello flessibile all’ingresso dell’ormone
Il gruppo ha scoperto una regione a sorpresa definita “cancello”, chiamata ansa extracellulare 2 (ECL2), che si trova in cima alla tasca di legame dell’ormone. Nello stato privo di farmaco (apo), questa ansa giace piatta sopra la tasca come un coperchio e non forma il tipico ponte disolfuro (un legame zolfo–zolfo) osservato in molti recettori correlati. Invece, parti dell’ansa si ripiegano nella tasca e sono tenute in posizione da una rete di interazioni con le eliche circostanti, coprendo parzialmente la grande cavità di legame adesiva. Quando uno dei tre antagonisti si lega, ECL2 si solleva e si allontana, forma il ponte disolfuro classico e crea una vasta cavità riempita di solvente che i farmaci occupano. Questo movimento drammatico suggerisce che V1a possa usare ECL2 come barriera dinamica per limitare l’attivazione casuale da molecole estranee, e che i farmaci potrebbero essere progettati per intrappolare l’ansa nello stato piatto “di base” o per sfruttarne la conformazione sollevata e aperta.
Come tre farmaci silenziano lo stesso recettore in modi diversi
Atosiban, che imita da vicino l’ormone naturale ossitocina, si estende dalla parte alta della tasca fino alla sua base, ancorandosi tramite una combinazione di legami a idrogeno e contatti idrofobici. Modificando poche posizioni chiave rispetto all’ossitocina, non riesce a innescare la catena di spostamenti interni normalmente necessari per l’attivazione del recettore: residui microswitch cruciali che si muovono durante la segnalazione restano bloccati nelle loro posizioni inattive, la cavità interna che accoglierebbe la proteina G non si apre e un sito di legame per il magnesio importante per l’attivazione viene perturbato. Al contrario, balovaptan e SRX246 sono molecole piccole, non peptidiche, che penetrano in profondità nella tasca ma impiegano strategie distinte. Balovaptan si affida a un nucleo rigido e idrofobico che si incastra strettamente in una fenditura profonda, oltre a una coda polare flessibile che si estende verso l’ingresso della tasca. SRX246 utilizza un’architettura modulare, simile a frammenti, ancorata da un nucleo β-lattamico, con diverse “zone” che tappano sottotasche ed estendono verso le anse extracellulari. In entrambi i casi, i farmaci stabilizzano una conformazione inattiva incompatibile con il legame della proteina G. Differenze sottili nella forma e nella chimica della tasca — specialmente in due posizioni sulle eliche 5 e 7 — aiutano a spiegare perché balovaptan e SRX246 preferiscano V1a rispetto a recettori strettamente correlati.
Implicazioni per terapie future
Fornendo istantanee ad alta risoluzione di V1a come dimero, rivelando una conformazione dell’ansa “piatta” nello stato privo di farmaco finora non vista e dettagliando come tre antagonisti molto diversi inattivano il recettore, questo lavoro fornisce ai progettisti di farmaci una mappa strutturale precisa per mirare a V1a. Suggerisce modi per creare medicinali di nuova generazione che sfruttino caratteristiche specifiche del dimero o blocchino il recettore in uno stato di base particolarmente inattivo, con l’obiettivo finale di trattare in modo più mirato i disturbi cerebrali e legati allo stress, riducendo gli effetti collaterali.
Citazione: Zhong, P., Chu, B., Yu, Z. et al. Molecular basis of antagonism of the dimeric human arginine vasopressin receptor 1A. Nat Commun 17, 1622 (2026). https://doi.org/10.1038/s41467-026-68331-5
Parole chiave: recettore della vasopressina V1a, recettore accoppiato a proteine G, dimerizzazione del recettore, struttura cryo-EM, progettazione di farmaci neuropsichiatrici