Clear Sky Science · it
Buone pratiche e strumenti in R e Python per l’elaborazione statistica e la visualizzazione dei dati di lipidomica e metabolomica
Perché trasformare i numeri di laboratorio in immagini chiare è importante
Gli strumenti moderni possono misurare migliaia di piccole molecole—lipidi e altri metaboliti—in una singola goccia di sangue o tessuto. Queste misurazioni contengono indizi sui rischi di malattia, sulle risposte ai trattamenti e su come il nostro organismo risponde a dieta o invecchiamento. Ma l’output grezzo non è una risposta pronta: è una grande tabella di numeri che va pulita, analizzata e trasformata in immagini comprensibili. Questo articolo spiega come i ricercatori possano usare due linguaggi di programmazione diffusi, R e Python, per farlo in modo affidabile, trasparente e con grafici di qualità pubblicabile.
Dalle misure chimiche a tabelle di dati complesse
Nella lipidomica e nella metabolomica, spettrometria di massa e cromatografia generano grandi set di dati in cui ogni riga è un campione e ogni colonna è una molecola. Queste tabelle raramente si comportano come esempi ordinati da manuale. Contengono valori mancanti, outlier e distribuzioni asimmetriche dove poche molecole mostrano livelli estremamente elevati. Le concentrazioni possono spaziare su diversi ordini di grandezza e possono essere influenzate da età, sesso, dieta, farmaci, ritmi giornalieri e problemi tecnici come deriva dello strumento o effetti di batch. Gruppi di esperti internazionali hanno emanato linee guida per standardizzare la raccolta, la lavorazione e la segnalazione dei campioni, ma anche con buone pratiche di laboratorio una accurata elaborazione statistica resta essenziale per estrarre segnali biologici veri da questo rumore di fondo.
Pulire e preparare i numeri
Prima che qualsiasi confronto tra gruppi sani e malati abbia senso, i dati vanno preparati. La recensione descrive come si generano i valori mancanti—per eventi casuali, limiti dello strumento o interferenze di segnale—e spiega quando possono essere ignorati in sicurezza, quando è necessario rimesurarli e come possono essere stimati in modo sensato (imputati) usando metodi come k-nearest neighbors, random forest o semplici sostituzioni con valori bassi. Successivamente, gli autori illustrano strategie di normalizzazione che riducono la variazione indesiderata, per esempio correggendo gli effetti di batch con campioni di controllo di qualità o adeguando le differenze nella quantità di campione. Discutono poi trasformazioni come il logaritmo—che addomestica code lunghe a destra nelle distribuzioni—e metodi di scaling che pongono tutte le molecole su una scala comparabile in modo che composti altamente variabili non dominino le analisi successive. 
Test statistici e narrazioni visive
Una volta che i dati sono preparati correttamente, entra in gioco un insieme di strumenti statistici. Per singole molecole i ricercatori possono calcolare fold change e usare test classici come il t-test o i suoi equivalenti non parametrici (ad esempio il test di Mann–Whitney) per chiedersi se i livelli differiscono tra gruppi. Per confronti che coinvolgono più gruppi si introducono metodi come ANOVA o il test di Kruskal–Wallis, accompagnati da procedure post hoc per individuare quali gruppi differiscono. La potenza di questi test si sblocca quando i risultati vengono visualizzati chiaramente. L’articolo evidenzia i box plot (incluse versioni migliorate per dati asimmetrici), i violin plot e i volcano plot che combinano dimensione dell’effetto e significatività statistica. Per i lipidi vengono descritte visualizzazioni più specializzate, come reti lipidiche che mostrano cambiamenti coordinati a livello di classi e grafici delle catene aciliche che rivelano pattern nella lunghezza e nella saturazione delle catene carboniose.
Scoprire pattern in molte variabili contemporaneamente
Poiché ogni campione può avere centinaia o migliaia di molecole misurate, i metodi multivariati sono cruciali. La recensione spiega come l’analisi delle componenti principali (PCA) comprima questa complessità in pochi nuovi assi che catturano le principali direzioni di variazione, consentendo controlli rapidi per separazione dei gruppi, effetti di batch o stabilità analitica. Metodi non lineari più avanzati, inclusi t-SNE e UMAP, possono rivelare cluster sottili e strutture nello spazio ad alta dimensione. Per situazioni in cui l’obiettivo è classificare i campioni—per esempio distinguere pazienti da controlli—gli autori descrivono approcci supervisionati basati su Partial Least Squares e la sua estensione ortogonale (PLS-DA e OPLS-DA). Questi metodi collegano profili molecolari alle etichette dei campioni, supportano la selezione delle caratteristiche e sono spesso riassunti con grafici di score, loading plot e curve ROC.
Kit di strumenti pratici in R e Python
Per aiutare i principianti a passare dalla teoria alla pratica, l’articolo esamina un vasto ecosistema di pacchetti software. In R, raccolte come tidyverse e tidymodels semplificano la gestione dei dati e la modellazione, mentre ggplot2 e pacchetti aggiuntivi come ggpubr, ggstatsplot e tidyplots rendono più semplice generare figure pronte per la pubblicazione. Librerie specializzate gestiscono PCA, clustering e modelli basati su PLS, e i pacchetti Bioconductor supportano heatmap complesse e grafici interattivi. In Python, pandas fornisce la gestione delle tabelle, mentre matplotlib, seaborn e plotly coprono la visualizzazione, e scikit-learn offre un ampio set di metodi multivariati. In tutto il testo gli autori enfatizzano esempi passo-passo resi disponibili in un GitBook di accompagnamento, così i lettori possono riprodurre i workflow e adattarli ai propri dati. 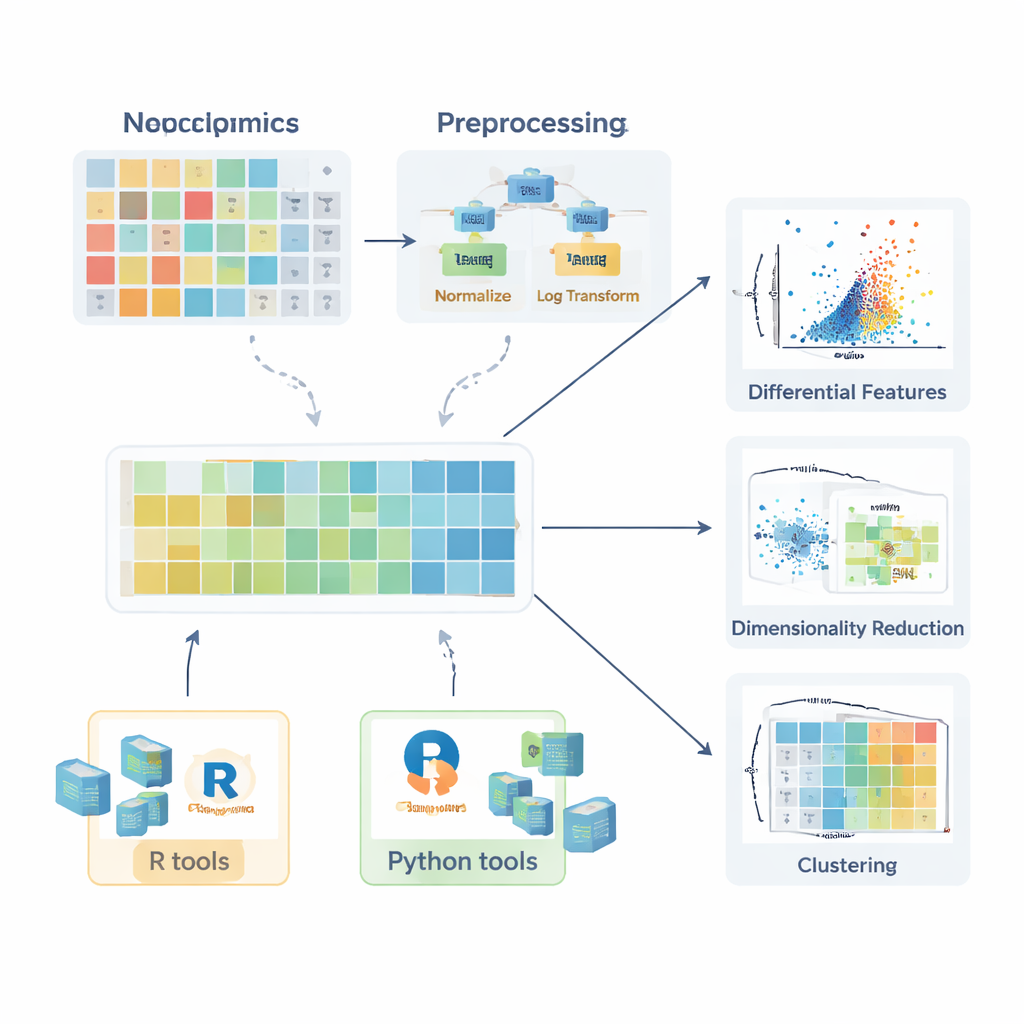
Trasformare chimica complessa in intuizioni affidabili
L’articolo conclude che la vera promessa della lipidomica e della metabolomica non risiede solo negli strumenti potenti, ma in quanto accuratamente il loro output viene elaborato e visualizzato. Seguendo buone pratiche statistiche, usando strumenti aperti e ben documentati in R e Python e facendo riferimento a esempi di codice condivisi, i ricercatori possono costruire pipeline robuste e riproducibili. Questo migliora le probabilità che i pattern trovati in molecole minime si traducano in biomarcatori attendibili, una migliore comprensione dei meccanismi della malattia e approcci più personalizzati alla medicina che, in ultima analisi, beneficiano i pazienti.
Citazione: Idkowiak, J., Dehairs, J., Schwarzerová, J. et al. Best practices and tools in R and Python for statistical processing and visualization of lipidomics and metabolomics data. Nat Commun 16, 8714 (2025). https://doi.org/10.1038/s41467-025-63751-1
Parole chiave: lipidomica, metabolomica, visualizzazione dei dati, programmazione R, Python