Clear Sky Science · it
Lo stato di metilazione della proteina fosfatasi 2A influenza l’alfa-sinucleinopatia nei modelli murini
Perché questo conta per la salute cerebrale
La malattia di Parkinson e i disturbi correlati sottraggono lentamente alle persone la capacità di muoversi, la memoria e l’indipendenza. Un colpevole principale è una proteina cerebrale chiamata alfa-sinucleina che può ripiegarsi in modo anomalo, aggregarsi e danneggiare i neuroni. Questo studio pone una domanda promettente: invece di attaccare direttamente la proteina, possiamo modulare i meccanismi di pulizia del cervello per impedire che l’alfa-sinucleina diventi tossica?
La storia di una proteina appiccicosa
Nella malattia di Parkinson e nella demenza a corpi di Lewy, grappoli contorti di alfa-sinucleina si accumulano all’interno dei neuroni, formando i classici “corpi di Lewy”. Un marcatore chimico particolare su questa proteina, aggiunto in un sito chiamato serina 129, è fortemente associato alla sua forma più dannosa. Quando questo marcatore è abbondante, l’alfa-sinucleina è più propensa a formare fibrille rigide e aggregati. Il cervello normalmente equilibra questi marcatori usando enzimi che li aggiungono e altri che li rimuovono. Poiché molti enzimi possono aggiungere il marcatore, bloccarne uno solo è improbabile che sia efficace. Gli autori si sono quindi concentrati sulla principale famiglia enzimatica che rimuove il marcatore, chiamata proteina fosfatasi 2A, o PP2A, che agisce come una cancellina molecolare per questa pericolosa modificazione.
La cancellina del cervello e i suoi due interruttori
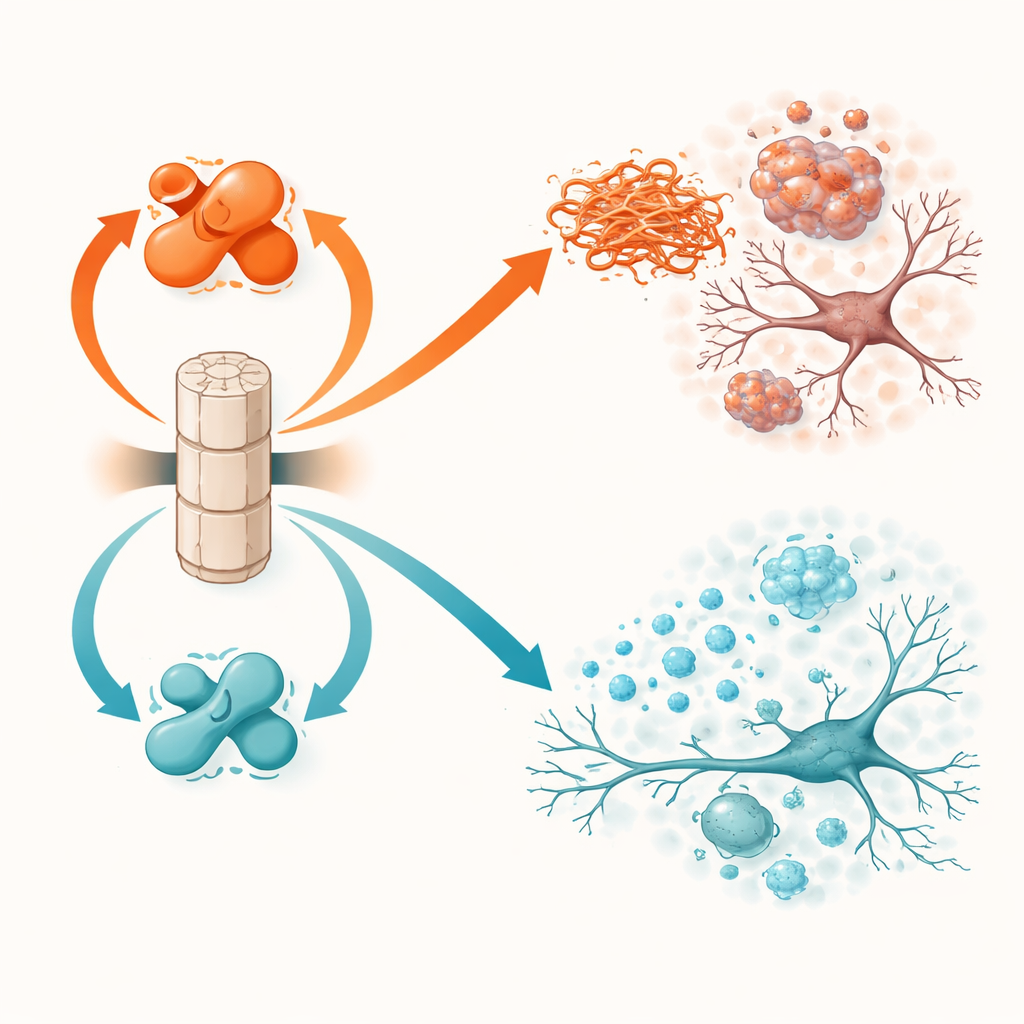
PP2A non funziona alla massima potenza di default. La sua attività dipende da una piccola modifica chimica, chiamata metilazione, su una delle sue subunità. Due altre proteine controllano questo interruttore: LCMT-1 aggiunge la modifica e orienta PP2A verso una forma più attiva e protettiva, mentre PME-1 la rimuove e spinge PP2A verso uno stato meno attivo e dannoso. Studi precedenti su tessuto cerebrale umano hanno mostrato che nella malattia di Parkinson e nella demenza a corpi di Lewy LCMT-1 tende a essere più bassa e PME-1 più alta, lasciando PP2A sottodimensionata. Lo studio attuale testa direttamente cosa accade quando questi interruttori vengono spinti deliberatamente in una direzione o nell’altra in topi viventi che sviluppano problemi di alfa-sinucleina.
Testare l’equilibrio nei cervelli viventi
I ricercatori hanno usato due modelli murini complementari. In uno, i topi sono stati ingegnerizzati per produrre alfa-sinucleina umana in tutto il cervello, sviluppando gradualmente aggregati proteici e problemi di movimento e memoria con l’età. Questi animali sono stati ulteriormente modificati per sovraprodurre o PME-1 (l’interruttore “off” di PP2A) o LCMT-1 (l’interruttore “on” di PP2A) nei neuroni del prosencefalo. Nel secondo modello, il team ha iniettato fibrille di alfa-sinucleina preformate nello striato, una regione profonda del cervello coinvolta nel movimento. Queste fibrille agiscono da semi, reclutando l’alfa-sinucleina normale e diffondendo la patologia nel corso di mesi in topi altrimenti normali o modificati per gli enzimi. In entrambi i modelli, gli scienziati hanno misurato l’accumulo proteico, la salute neuronale, l’infiammazione cerebrale e il comportamento.
Quando la cancellina vacilla, il danno si propaga
I topi che sovraproducevano PME-1, e quindi avevano PP2A meno attiva, andarono peggio. Negli animali transgenici per l’alfa-sinucleina, l’aumento di PME-1 portò a una maggiore presenza di alfa-sinucleina marcata e aggregata nella corteccia e nell’ippocampo, a una perdita più consistente della struttura neuronale, a segnali di attività neuronale più deboli e a una forte attivazione delle cellule immunitarie nel cervello. Questi cambiamenti si tradussero in prestazioni peggiori nei test di movimento e nei compiti di apprendimento e memoria. Nel modello di iniezione di fibrille, la sovraespressione di PME-1 permise l’accumulo e la diffusione più estesa di assemblaggi tossici di alfa-sinucleina, soprattutto nelle cellule nervose produttrici di dopamina della substantia nigra, una regione chiave persa nella malattia di Parkinson. Questi topi mostrarono una perdita più severa delle fibre dopaminergiche, un’infiammazione più intensa e maggiori deficit motori e di costruzione del nido.
Riacciuffare la cancellina
La manipolazione opposta, la sovrapproduzione di LCMT-1 per mantenere PP2A fortemente metilata e attiva, ebbe effetti ampiamente protettivi. Nei topi transgenici per l’alfa-sinucleina, LCMT-1 ridusse il carico di proteina marcata e aggregata fino a livelli prossimi alla normalità e preservò sia la struttura sia l’attività dei neuroni. I marcatori infiammatori risultarono più bassi e gli animali si comportarono in modo più simile ai controlli sani nei test di equilibrio e memoria. Nel modello di semina con fibrille, LCMT-1 limitò sia l’accumulo locale sia la diffusione a lunga distanza dell’alfa-sinucleina tossica, risparmiò i neuroni dopaminergici dalla degenerazione, ridusse l’attivazione delle microglia e attenuò il declino nella coordinazione motoria e nel comportamento di costruzione del nido. In tutti gli esperimenti, spostare PP2A verso il suo stato attivo e metilato si tradusse costantemente in benefici molecolari e in protezione funzionale.
Cosa potrebbe significare per i trattamenti futuri
Per i non specialisti, la lezione è semplice: il cervello ha una cancellina incorporata che può rimuovere un marcatore dannoso dall’alfa-sinucleina e impedire che si trasformi in aggregati pericolosi. Quando questa cancellina è indebolita, danno, infiammazione e sintomi peggiorano; quando è rafforzata, i neuroni sono protetti. Lo studio fornisce prove dirette in animali vivi che lo stato di metilazione di PP2A è un controllo maestro della tossicità dell’alfa-sinucleina e delle sue conseguenze. Questo indica una nuova strategia terapeutica: invece di inseguire ogni forma nociva della proteina, si potrebbero progettare farmaci per spingere PP2A e i suoi regolatori LCMT-1 e PME-1 verso uno stato più protettivo. Tali approcci richiederanno accurati test di sicurezza, ma promettono di rallentare o prevenire la malattia di Parkinson e condizioni correlate ristabilendo la capacità del cervello di tenere sotto controllo l’alfa-sinucleina.
Citazione: Maddila, S., Hassanzadeh, K., Liu, J. et al. Protein phosphatase 2A methylation state impacts α-synucleinopathy in mouse models. Cell Death Discov. 12, 172 (2026). https://doi.org/10.1038/s41420-026-03045-7
Parole chiave: Malattia di Parkinson, alfa-sinucleina, proteina fosfatasi 2A, neurodegenerazione, infiammazione cerebrale