Clear Sky Science · it
Varianti perdita di funzione in HPDL compromettono lo sviluppo corticale umano tramite alterazioni della funzione mitocondriale
Perché i piccoli motori cellulari sono importanti per il cervello in crescita
La maggior parte delle persone, pensando allo sviluppo cerebrale, immagina geni e circuiti. Questo studio mostra che un altro fattore, spesso trascurato — le piccole centrali energetiche all’interno delle nostre cellule chiamate mitocondri — può anch’esso influenzare la formazione del cervello. Studiano rari disordini del movimento dell’infanzia legati al gene HPDL, gli autori rivelano come una produzione di energia difettosa possa ridurre la corteccia in via di sviluppo, la regione cerebrale cruciale per movimento, pensiero e comportamento.
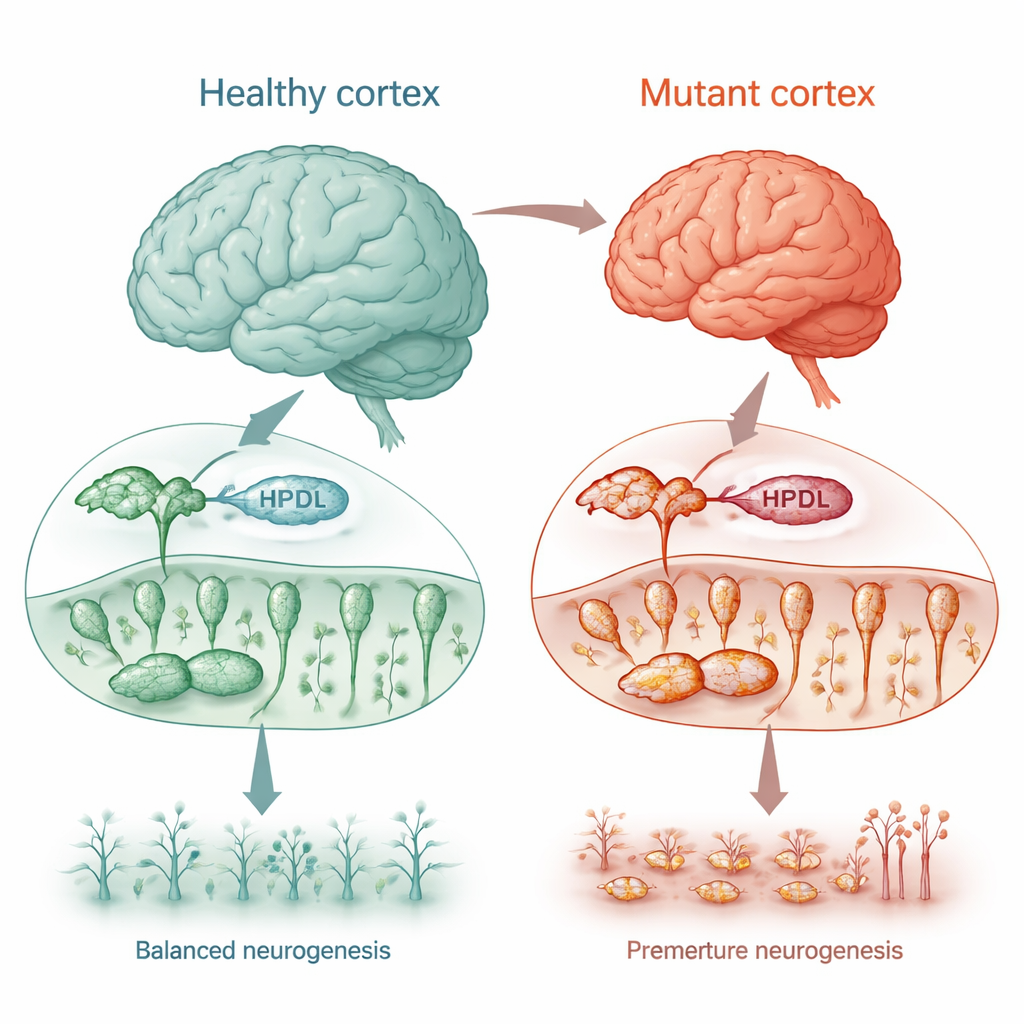
Un raro disturbo del movimento come finestra sulla crescita cerebrale
Alcuni bambini con alterazioni del gene HPDL sviluppano una spasticità ereditaria degli arti inferiori, una condizione che causa rigidità e debolezza delle gambe, insieme a crisi epilettiche, ritardo dello sviluppo e, nei casi più gravi, un cervello più piccolo del normale (microcefalia). Sebbene fosse noto che la proteina HPDL si localizza nei mitocondri, il suo ruolo esatto — e il motivo per cui la sua perdita danneggia il cervello — non era chiaro. I ricercatori hanno usato diversi modelli cellulari umani, comprese cellule tumorali di tipo neuronale e cellule cerebrali ricavate da campioni di cute dei pazienti, per verificare se HPDL sia necessario per lo sviluppo cerebrale normale e per la salute mitocondriale.
Cosa succede quando HPDL viene spento
Per prima cosa il gruppo ha disattivato HPDL in una linea cellulare umana di neuroblastoma usando l’editing genico CRISPR. Senza HPDL, queste cellule perdevano la proteina a lunghezza completa e manifestavano evidenti problemi mitocondriali. I grandi complessi della catena respiratoria che normalmente collaborano per generare energia risultavano disturbati, e componenti chiave coinvolti nell’uso dell’ossigeno risultavano ridotti. Le cellule consumavano meno ossigeno, producevano una respirazione legata all’energia inferiore e generavano più specie reattive dell’ossigeno — sottoprodotti dannosi spesso indicati come “stress ossidativo”. Tuttavia il numero totale di mitocondri non diminuiva, e i livelli di coenzima Q10, una molecola vitale per il trasferimento di energia, erano in realtà più alti, suggerendo un difetto di qualità — non solo quantitativo — nella funzione mitocondriale.
Il tessuto cerebrale in coltura rivela una sovrapproduzione precoce di neuroni
Per vedere come la perdita di HPDL influenzi il vero sviluppo cerebrale umano, i ricercatori hanno riprogrammato cellule cutanee di quattro bambini colpiti in cellule staminali pluripotenti indotte e le hanno poi indotte a formare cellule corticali e “mini-cervelli” tridimensionali (organoidi). Nelle fasi iniziali dello sviluppo, a uno stadio in cui la maggior parte delle cellule dovrebbe ancora dividersi come progenitori neurali, le colture con mutazioni in HPDL contenevano già più neuroni maturi e meno progenitori. I profili di espressione genica confermavano questo dato: le vie che guidano la formazione neuronale venivano attivate troppo presto, mentre quelle che mantengono le cellule in stato di proliferazione risultavano attenuate. Negli organoidi, questo passaggio prematuro da elementi costitutivi a neuroni maturi portò a strutture cerebrali molto più piccole, riecheggiando la microcefalia osservata nei bambini più gravemente colpiti.
Centrali energetiche rotte e cellule sotto stress
Un’ispezione più dettagliata mostrò che le cellule cerebrali mutanti per HPDL avevano un’ossidativa fosforilazione compromessa — il principale meccanismo con cui i mitocondri producono energia. Colorazioni enzimatiche rivelarono un’attività ridotta di un complesso mitocondriale chiave, mentre altre misure mostrarono un’alterata carica elettrica attraverso la membrana mitocondriale. In molte cellule mutanti, un enzima cruciale che normalmente sintetizza ATP sembrava funzionare al contrario per sostenere questa carica di membrana, segno di un profondo disagio metabolico. Tra le linee dei pazienti, le specie reattive dell’ossigeno erano costantemente elevate e i grandi complessi della catena respiratoria si formavano meno adeguatamente. Questi cambiamenti mitocondriali seguivano da vicino i tempi e il grado di produzione neuronale prematura.
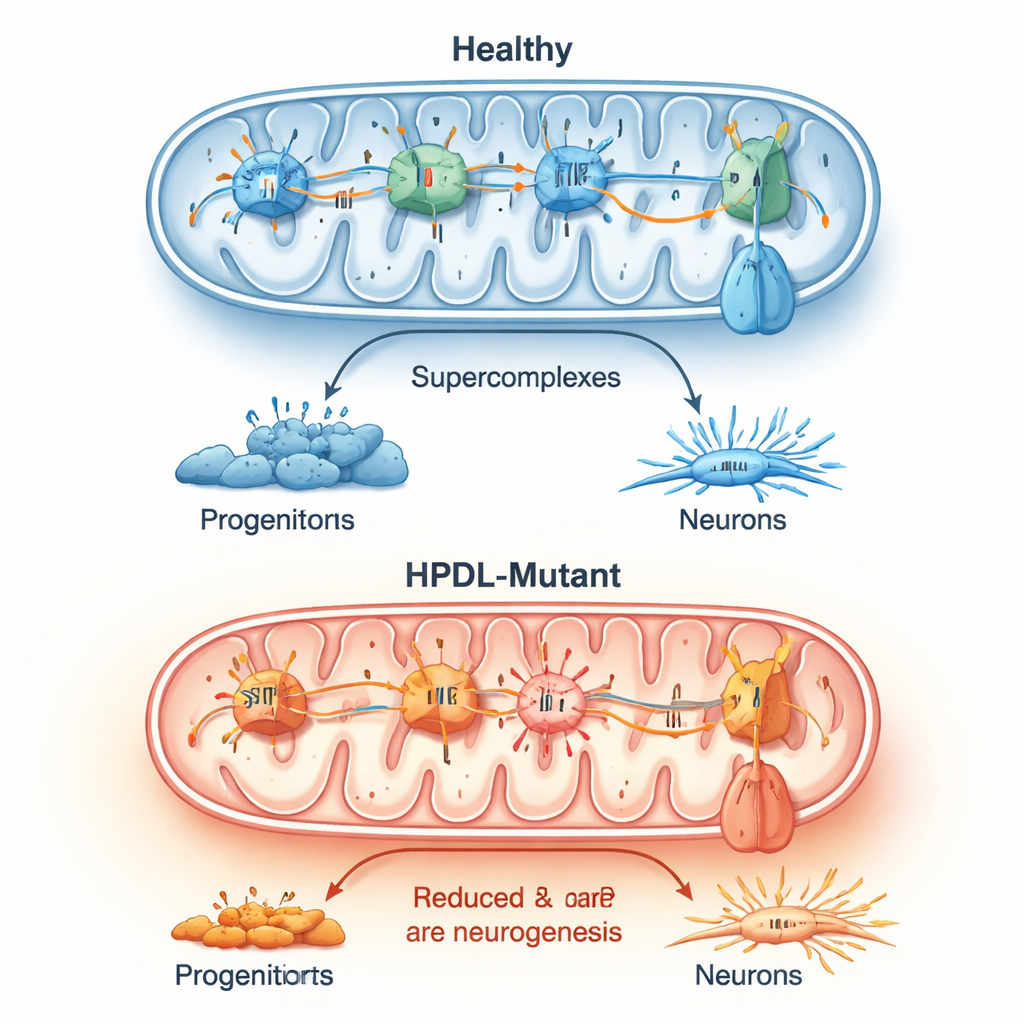
Testare modi per alleviare lo stress
Dato che lo stress ossidativo e la chimica alterata del coenzima Q10 sembravano centrali, il team ha testato se trattamenti rivolti a questi problemi potessero rallentare il passaggio verso la formazione neuronale. Hanno esposto colture corticali precoci a due antiossidanti e a 4-idrossibenzoato, una piccola molecola correlata alla sintesi del coenzima Q10. In diverse linee derivate dai pazienti, questi composti ridussero parzialmente la neurogenesi prematura, ma la risposta dipese dalla specifica mutazione di HPDL. Alcune linee risposero principalmente agli antiossidanti, altre al precursore del coenzima Q10, e una non rispose affatto. Questo schema dipendente dalla mutazione suggerisce che per i disturbi correlati a HPDL potrebbero servire strategie di trattamento personalizzate.
Cosa significa per i bambini e per le terapie future
In termini semplici, questo studio mostra che HPDL agisce come guardiano dei mattoni del cervello durante lo sviluppo precoce. Quando HPDL fallisce, i mitocondri diventano inefficienti eccessivamente sotto stress, spingendo i progenitori a trasformarsi in neuroni troppo presto. La riserva di cellule in divisione si esaurisce, la corteccia non raggiunge la sua piena dimensione e gli schemi di connessione vengono alterati, contribuendo a problemi di movimento e ad altri sintomi. Il recupero parziale osservato con antiossidanti e composti correlati al coenzima Q10 lascia intendere che modulare l’equilibrio energetico cellulare e lo stress ossidativo potrebbe un giorno aiutare i bambini con mutazioni in HPDL, e forse anche altri con forme mitocondriali di malattia cerebrale.
Citazione: Baggiani, M., Desbats, M.A., Naef, V. et al. Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function. Cell Death Dis 17, 237 (2026). https://doi.org/10.1038/s41419-026-08476-9
Parole chiave: HPDL, mitocondri, sviluppo corticale, microcefalia, stress ossidativo