Clear Sky Science · it
La TDP-43 mutante causa deficit nel trasporto assonale e nella glicolisi in un modello di neuroni motori derivati da cellule staminali murine della sclerosi laterale amiotrofica (SLA)
Perché questa ricerca è importante per le persone con SLA
La sclerosi laterale amiotrofica (SLA) è una malattia fatale che paralizza progressivamente le persone distruggendo i neuroni che controllano i muscoli. Nella maggior parte dei pazienti si osservano alterazioni che coinvolgono una proteina chiamata TDP-43, ma gli scienziati non comprendono ancora completamente come tali alterazioni danneggino i motoneuroni. Questo studio utilizza un modello murino di cellule staminali accuratamente progettato per identificare alcuni dei problemi precoci causati da una versione della TDP-43 associata alla malattia, offrendo indizi che potrebbero orientare futuri trattamenti.
Costruire neuroni motori in provetta
Per esplorare la SLA in condizioni controllate, i ricercatori hanno iniziato con cellule staminali embrionali murine e le hanno indirizzate a diventare neuroni motori, le cellule lunghe e simili a cavi che trasmettono segnali dal midollo spinale ai muscoli. Hanno inserito una copia aggiuntiva del gene umano TDP-43 in queste cellule, sia nella forma normale sia portatrice di una specifica mutazione associata alla SLA chiamata M337V. Un marcatore fluorescente ha permesso al team di seguire la proteina umana all’interno delle cellule. Al giorno 20 di coltura, sia le cellule con la proteina normale sia quelle con la proteina mutante erano maturate in neuroni motori che esprimevano marcatori tipici, formavano reti ramificate e stabilivano connessioni simili a sinapsi, riproducendo da vicino i neuroni del sistema nervoso. 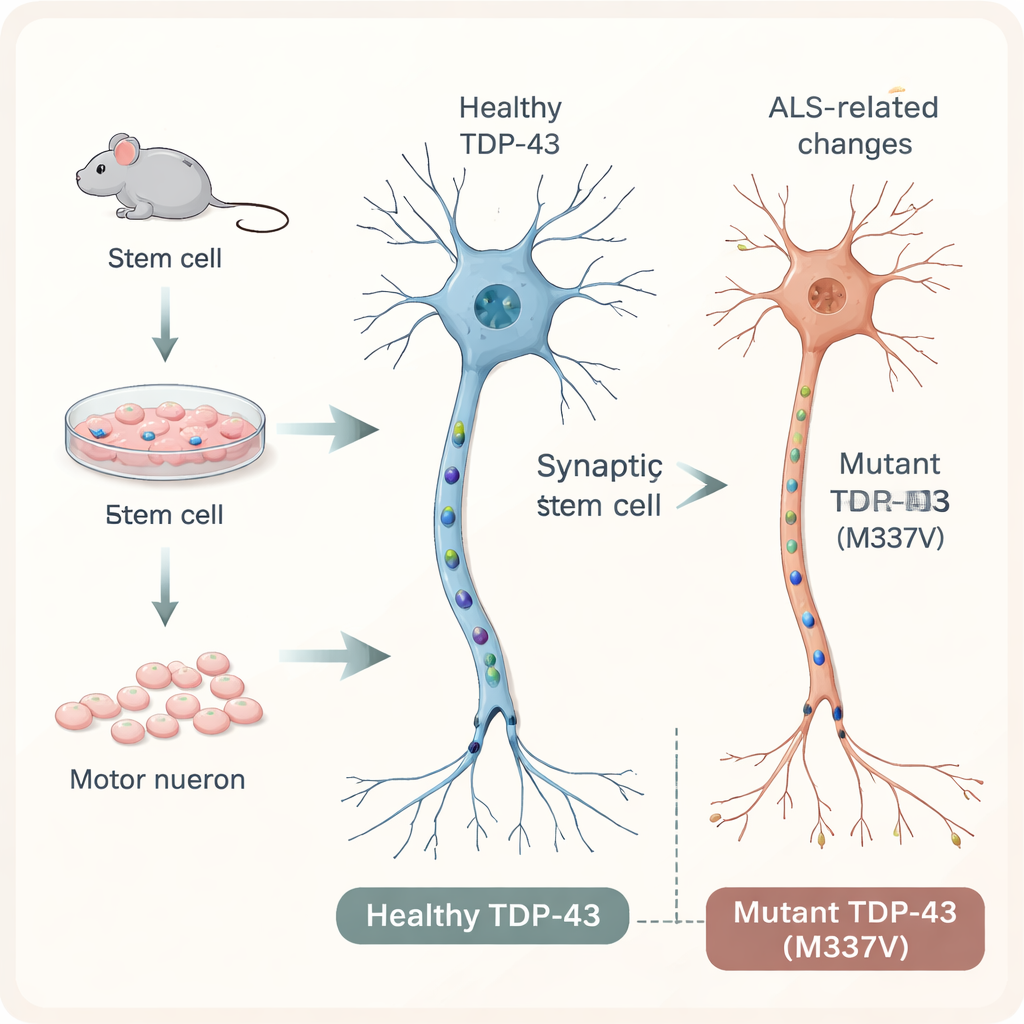
Danno nascosto senza aggregati proteici visibili
Nelle persone con SLA, la TDP-43 spesso si sposta dalla sua posizione abituale nel nucleo al citoplasma e forma aggregati, un classico segno patologico osservabile al microscopio. Sorprendentemente, in questo modello di cellule staminali la TDP-43 mutante non mostrava una aumentata mislocalizzazione o aggregazione rispetto alla versione normale. La maggior parte della proteina rimaneva nel luogo previsto. Eppure i neuroni erano chiaramente meno sani: le colture con la proteina mutante presentavano meno corpi cellulari, reti di fibre nervose più ridotte e una sopravvivenza complessiva inferiore. Ciò suggerisce che danni significativi ai motoneuroni possono manifestarsi prima, o anche senza, i drammatici aggregati proteici osservati nei cervelli e nei midolli spinali dei pazienti.
Ingorgo sulle “autostrade” neuronali
I motoneuroni dipendono da sistemi di trasporto rapidi ed efficienti che spostano carichi vitali lungo i loro lunghi assoni. Utilizzando dispositivi microfluidici che isolano gli assoni in canali microscopici, il team ha seguito il movimento di vescicole di segnalazione (endosomi) e dei mitocondri produttori di energia in cellule vive. Nei neuroni con TDP-43 mutante, questi carichi si muovevano ancora per lo più nelle direzioni corrette, ma a velocità ridotta. Gli endosomi di segnalazione mostravano velocità ridotte nel trasporto retrogrado verso il corpo cellulare, e i mitocondri si muovevano più lentamente in entrambe le direzioni, con periodi di pausa più frequenti. È importante notare che il macchinario di base che guida questo movimento—le proteine motrici che camminano lungo i binari intracellulari—non sembrava cambiare in quantità, suggerendo che il problema riguarda il funzionamento di questo sistema più che la sua abbondanza.
Carente energia dalla glicolisi, ma non dai mitocondri
Poiché il trasporto assonale richiede molta energia, i ricercatori hanno testato come questi neuroni producono e utilizzano energia. Hanno misurato due fonti principali: la respirazione mitocondriale, che brucia combustibile usando ossigeno, e la glicolisi, che scompone lo zucchero nel fluido circostante. I mitocondri apparivano normali per numero, forma e potenziale di membrana, e la loro capacità complessiva di produrre energia sembrava non modificata nelle cellule mutanti. Al contrario, i neuroni con TDP-43 mutante mostravano una chiara riduzione della glicolisi basale. Lavori precedenti hanno dimostrato che la glicolisi locale lungo gli assoni può fornire carburante “a bordo” per il rapido trasporto di vescicole. La ridotta capacità di bruciare zucchero osservata qui potrebbe quindi contribuire al rallentamento del movimento dei carichi, aggiungendo un ulteriore stress a motoneuroni già vulnerabili.
Cosa significa per le terapie future della SLA
Nel complesso, lo studio mostra che anche bassi livelli di TDP-43 mutante associata alla SLA sono sufficienti a rendere i neuroni motori più fragili, rallentare il movimento dei carichi essenziali lungo gli assoni e attenuare la loro capacità di generare energia dallo zucchero—il tutto senza i chiari aggregati proteici che i patologi solitamente cercano. Per i non specialisti, il messaggio chiave è che cambiamenti precoci e sottili nel “flusso del traffico” cellulare e nell’uso dell’energia possono predisporre a danni successivi e più gravi nella SLA. Questo evidenzia il trasporto assonale e le vie energetiche cellulari, in particolare la glicolisi, come bersagli promettenti per terapie mirate a proteggere i motoneuroni prima che si verifichi una degenerazione irreversibile. 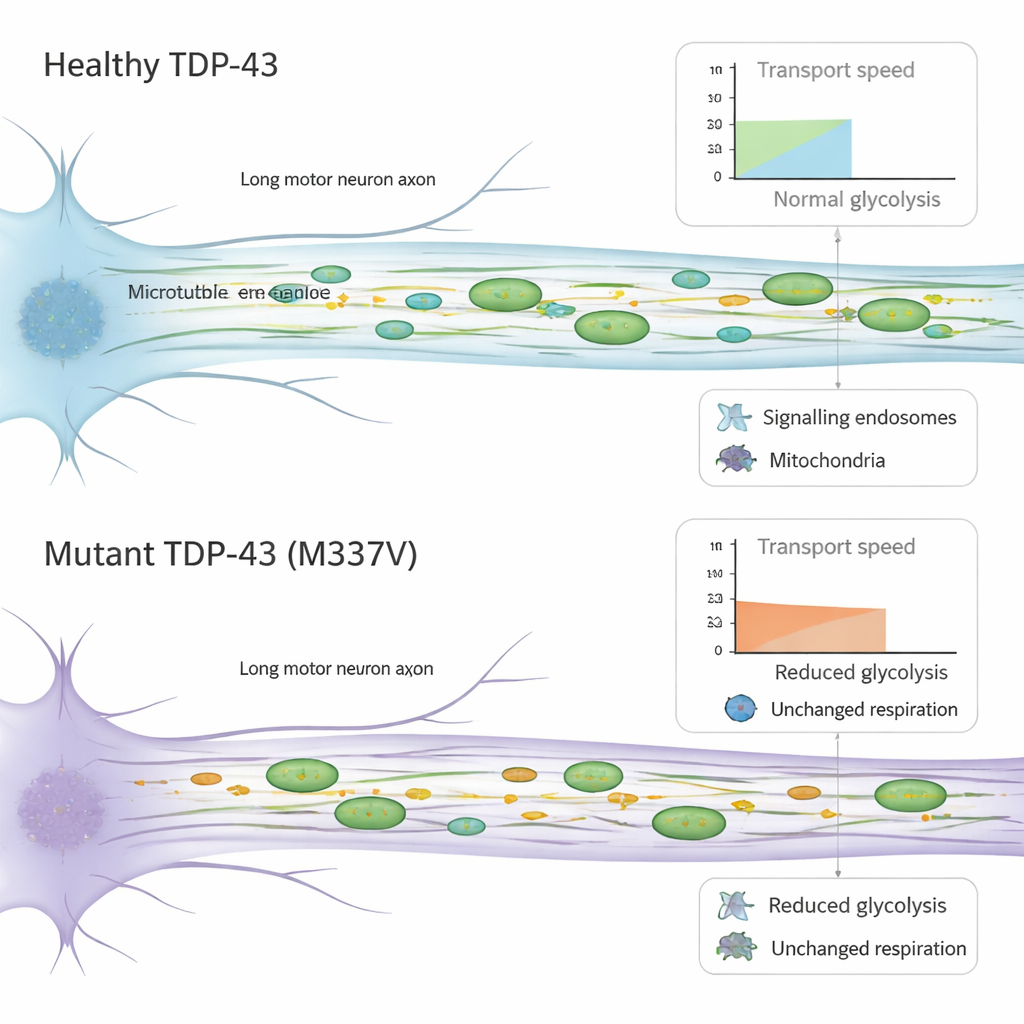
Citazione: Carroll, E., Scaber, J., Pasniceanu, IS. et al. Mutant TDP-43 drives impairments in axonal transport and glycolysis in a mouse stem-cell-derived motor neuron model of amyotrophic lateral sclerosis (ALS). Cell Death Dis 17, 193 (2026). https://doi.org/10.1038/s41419-026-08437-2
Parole chiave: sclerosi laterale amiotrofica, TDP-43, neuroni motori, trasporto assonale, energia cellulare