Clear Sky Science · it
La disfunzione degli interneuroni gabaergici è alla base delle oscillazioni di rete neuronale alterate associate ad attività epilettiforme nei topi privi di PPT1
Quando i ritmi cerebrali si guastano
Le crisi non sono soltanto improvvise tempeste di attività cerebrale; spesso nascono da cambiamenti sottili nel modo in cui le cellule nervose comunicano. Questo studio esamina una rara malattia cerebrale infantile, la malattia CLN1, e pone una domanda semplice ma di ampia portata: cosa succede ai “regolatori del ritmo” del cervello quando manca un solo enzima, chiamato PPT1? Seguendo queste modificazioni nei topi nel tempo, i ricercatori mostrano come piccoli difetti precoci nell’inibizione possano espandersi fino a provocare crisi e danni cerebrali diffusi.
I guardiani dell’equilibrio cerebrale
I nostri cervelli si affidano a due grandi tipi di cellule nervose. Le cellule eccitatorie, come i neuroni piramidali nell’ippocampo, promuovono l’attività; le cellule inibitorie, chiamate interneuroni, fanno da freno, contenendo quell’attività e plasmando i ritmi elettrici del cervello. Tra questi, due gruppi importanti sono gli interneuroni parvalbumina-positivi (PV+) e quelli somatostatina-positivi (SST+). Essi contribuiscono a generare e coordinare onde cerebrali ritmiche, come le oscillazioni theta e gamma, che sostengono funzioni quali apprendimento e memoria. Nella malattia CLN1, i bambini perdono l’enzima PPT1, che normalmente rimuove gruppi grassi dalle proteine. Gli autori hanno usato un modello murino con la stessa mutazione riscontrata nei pazienti per vedere come questa perdita influenzi gli interneuroni e i ritmi cerebrali che essi aiutano a controllare.
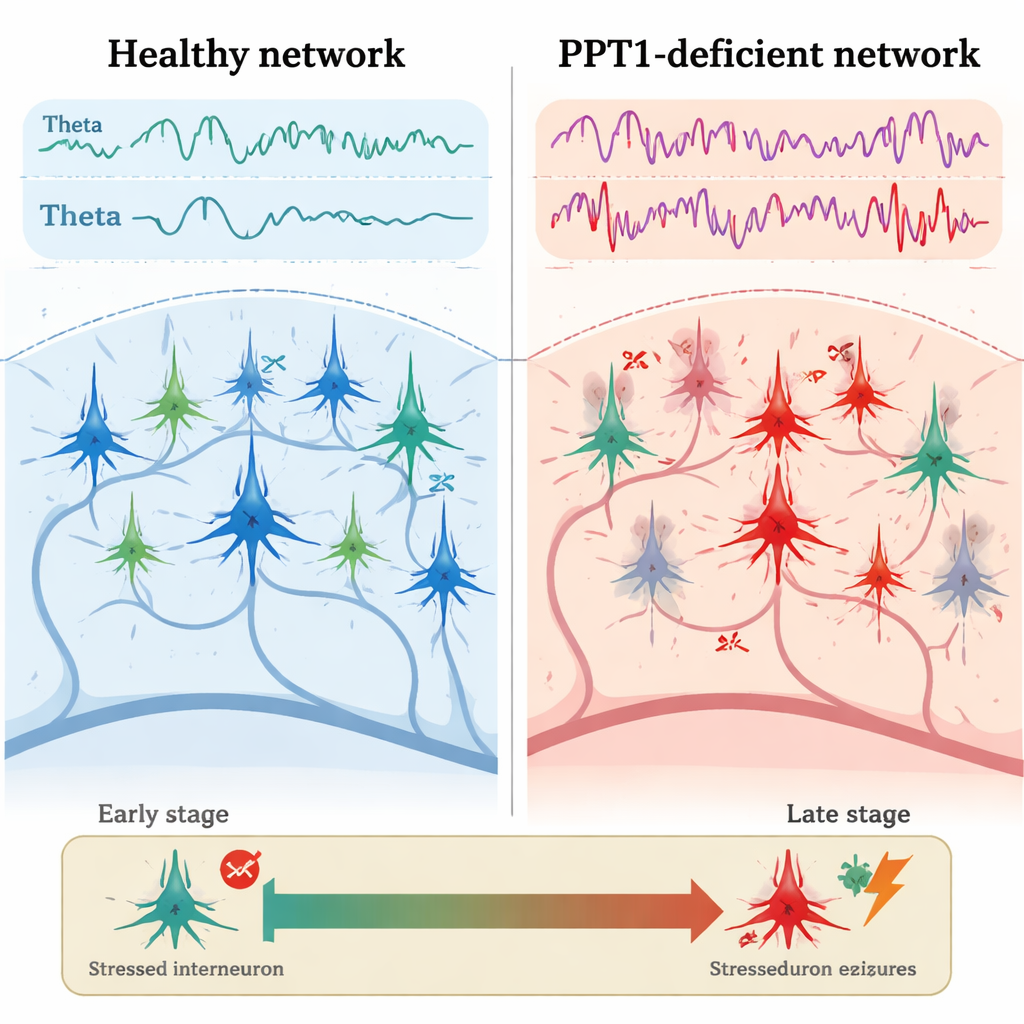
Prime crepe nel sistema inibitorio
Nei topi mutanti giovani-adulti, di circa tre-quattro mesi, il primo problema netto è emerso negli interneuroni PV+. Registrazioni elettriche dall’ippocampo hanno mostrato che queste cellule inibitorie scaricavano con minore frequenza rispetto ai topi sani, mentre i neuroni piramidali vicini sparavano più rapidamente e con pause più brevi tra gli spike. La microscopia ha rivelato che molti interneuroni PV+ presentavano caspasi-3 attivata, un importante esecutore della morte cellulare programmata, nonostante il loro numero complessivo non fosse ancora diminuito. Allo stesso tempo, la potenza delle onde theta e gamma era aumentata, e l’imaging del calcio mostrava un’attività più intensa nei neuroni ippocampali mentre gli animali si muovevano. Elemento cruciale, il normale “dialogo” tra theta e gamma — dove le onde più lente organizzano quelle più rapide — risultava indebolito, suggerendo un precoce deterioramento del fine timing dell’attività di rete.
Da ritmi disturbati a scariche epilettiche
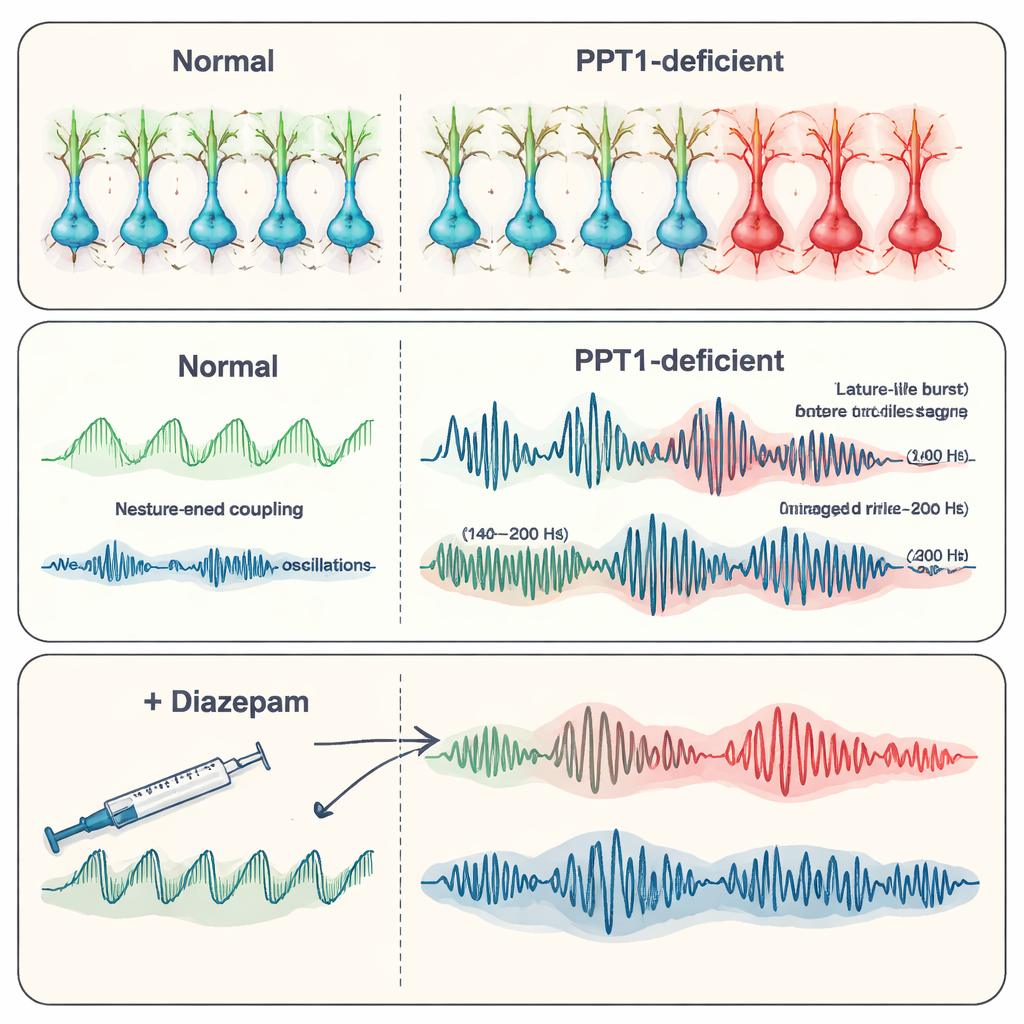
Entro i sei-sette mesi di età, il quadro è peggiorato. Molti interneuroni PV+ erano andati persi, e anche gli interneuroni SST+ mostravano segni di attivazione della caspasi-3. Le registrazioni dall’ippocampo hanno rivelato scariche epilettiformi spontanee — brevi esplosioni anomale di attività associate alle crisi. Il gruppo si è concentrato sui “ripple” ad alta frequenza, oscillazioni veloci che normalmente aiutano l’immagazzinamento della memoria. Nei topi mutanti, i ripple fisiologici (intorno a 140–200 hertz) sono diventati meno frequenti ma di ampiezza maggiore, mentre ripple ancora più veloci e “patologici” (200–500 hertz), strettamente legati all’epilessia, sono cresciuti in forza e frequenza. Complessivamente, queste modifiche suggeriscono uno spostamento dai ritmi organizzati e legati alla memoria verso pattern caotici e proclivi alle crisi man mano che il controllo inibitorio viene meno.
I neuroni si logorano e il diazepam interviene
Con l’avanzare della malattia, l’ippocampo stesso ha iniziato a degenerare. I segnali di calcio nei neuroni sono diminuiti, la colorazione di Golgi ha mostrato alberi dendritici più sottili e meno ramificati, e sono risultate ridotte le spine sinaptiche dove si formano le connessioni. I conteggi dei neuroni nelle regioni ippocampali chiave (CA1 e CA3) hanno confermato una perdita cellulare diffusa, e nelle registrazioni elettriche si captavano meno unità attive. I ricercatori hanno quindi testato il diazepam, un comune farmaco anticonvulsivante che potenzia l’azione dell’inibitorio GABA. Nei topi mutanti più anziani, il diazepam ha ridotto la frequenza delle scariche epilettiche e ha parzialmente ripristinato pattern oscillatori più normali, inclusi i ripple, sebbene non abbia riparato la perdita sottostante di recettori. Ciò suggerisce che rafforzare i segnali inibitori residui può comunque calmare la rete, almeno temporaneamente.
Perché questi risultati contano
Per il lettore non specialistico, il messaggio chiave è che la malattia CLN1 non è solo accumulo di rifiuti all’interno delle cellule cerebrali. La perdita di PPT1 innesca una reazione a catena: prima, interneuroni inibitori specializzati si stressano e cominciano a fallire, liberando neuroni piramidali iperattivi e distorcendo i ritmi cerebrali. Col tempo, questo squilibrio conduce a crisi e infine a una perdita su larga scala di cellule e connessioni cerebrali. Lo studio individua una finestra di opportunità precoce nella malattia, quando proteggere o salvare gli interneuroni PV+ — per esempio bloccando l’attivazione delle caspasi — potrebbe prevenire crisi e degenerazione successive. Pur non potendo curare la CLN1, la capacità del diazepam di attenuare i ritmi anomali in questo modello sottolinea l’idea più ampia che ripristinare l’inibizione potrebbe essere una strategia efficace nell’epilessia e in disturbi cerebrali correlati.
Citazione: Tong, J., Liu, W., Wang, Q. et al. Dysfunction of GABAergic interneurons underlies altered neural network oscillations associated with epileptiform activity in PPT1-deficient mice. Transl Psychiatry 16, 106 (2026). https://doi.org/10.1038/s41398-026-03843-8
Parole chiave: epilessia, interneuroni, ippocampo, oscillazioni cerebrali, malattia da accumulo lisosomiale