Clear Sky Science · it
Potenziare l’attività di KLF15 nei cardiomiociti: un approccio innovativo per prevenire il riprogrammamento patologico e la fibrosi tramite dCas9VPR privo di nucleasi
Riprogrammare il cuore in insufficienza
Lo scompenso cardiaco colpisce milioni di persone, spesso sviluppandosi lentamente dopo anni di ipertensione o malattia valvolare. In queste condizioni, le cellule del muscolo cardiaco non solo aumentano di volume, ma riattivano un programma genetico «fetale» e il cuore si riempie di tessuto cicatriziale. Questo studio esplora un nuovo modo per riportare il controllo genico del cuore verso la normalità — senza tagliare il DNA — aumentando delicatamente un regolatore protettivo chiamato KLF15 nei cardiomiociti.
Quando le cellule cardiache perdono la propria identità
In un cuore adulto sano, i cardiomiociti — le cellule del muscolo cardiaco — bruciano grassi in modo efficiente per produrre energia e mantengono uno schema stabile di espressione genica. Utilizzando l’RNA-seq a singola cellula in topi sottoposti a sovraccarico di pressione cronico, i ricercatori hanno mappato come singoli cardiomiociti cambiano mentre il cuore passa da funzione normale a dilatazione e infine a insufficienza. Hanno scoperto che un fattore di trascrizione chiamato KLF15, che normalmente equilibra metabolismo e crescita, mostrava la variazione di attività più marcata nelle cellule malate. Con l’aumentare dello stress, i livelli di KLF15 diminuivano e la sua capacità di reprimere geni fetali e legati allo stress si indeboliva. Cadute simili di KLF15 sono state osservate anche in cuori umani di pazienti con cardiomiopatia dilatativa e ipertrofica, indicando che questa disfunzione è conservata tra le specie.
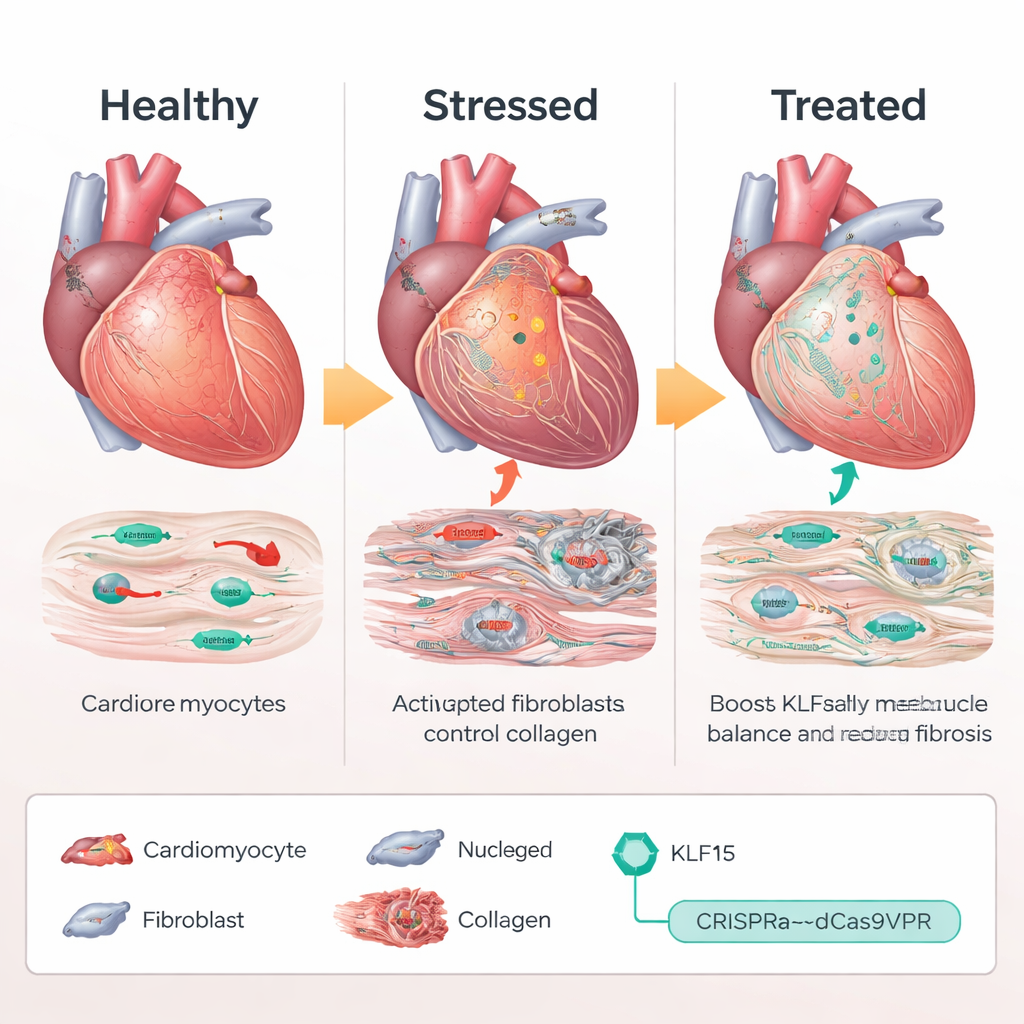
Usare CRISPR come un controllo del volume, non come forbici
Piuttosto che aggiungere una copia extra del gene KLF15 o tagliare il DNA, il gruppo ha utilizzato un sistema basato su CRISPR di «attivazione», chiamato dCas9VPR, che si lega vicino al gene Klf15 naturale e ne aumenta l’espressione. In topi ingegnerizzati per esprimere questo attivatore CRISPR solo nei cardiomiociti, gli scienziati hanno somministrato RNA guida tramite un virus adeno-associato (AAV9) per indirizzare il promotore di Klf15. Sottoposti a sovraccarico di pressione cronico, i topi che ricevevano guide attivatrici di Klf15 mantennero livelli quasi normali di Klf15. I loro cardiomiociti rimasero più piccoli, la funzione di pompaggio si ridusse meno e la sopravvivenza migliorò rispetto agli animali controllo. A livello molecolare, i geni dello stress e quelli fetali si sopirono, mentre i geni metabolici e quelli coinvolti nella gestione del calcio ripresero funzione, indicando che il programma trascrizionale patologico era stato sostanzialmente ripristinato.
Attenuare la formazione di cicatrice attraverso il dialogo cellula-cellula
Lo scompenso cardiaco è guidato non solo da cardiomiociti malati ma anche da fibroblasti, cellule di supporto che producono collagene e formano tessuto cicatriziale rigido. Analisi a singola cellula e imaging tissutale mostrarono che il ripristino di Klf15 nei cardiomiociti riduceva l’attivazione dei fibroblasti e la fibrosi complessiva, nonostante la terapia genica non avesse mai preso di mira direttamente i fibroblasti. Il gruppo ha ricondotto questo effetto a una proteina secreta chiamata AZGP1. Quando Klf15 veniva potenziato nei cardiomiociti, aumentava la produzione e il rilascio di AZGP1. Nei cuori di topo e nei tessuti cardiaci umani derivati da cellule staminali, livelli più alti di AZGP1 attenuavano la via TGF-β / SMAD nei fibroblasti — un driver chiave della cicatrizzazione — abbassando i livelli di marcatori come α-SMA e POSTN. È importante notare che la sovraespressione di AZGP1 nei soli cardiomiociti non riprogrammava le cellule muscolari, dimostrando che KLF15 protegge primariamente i cardiomiociti in modo diretto e usa AZGP1 come messaggero per contenere i fibroblasti.
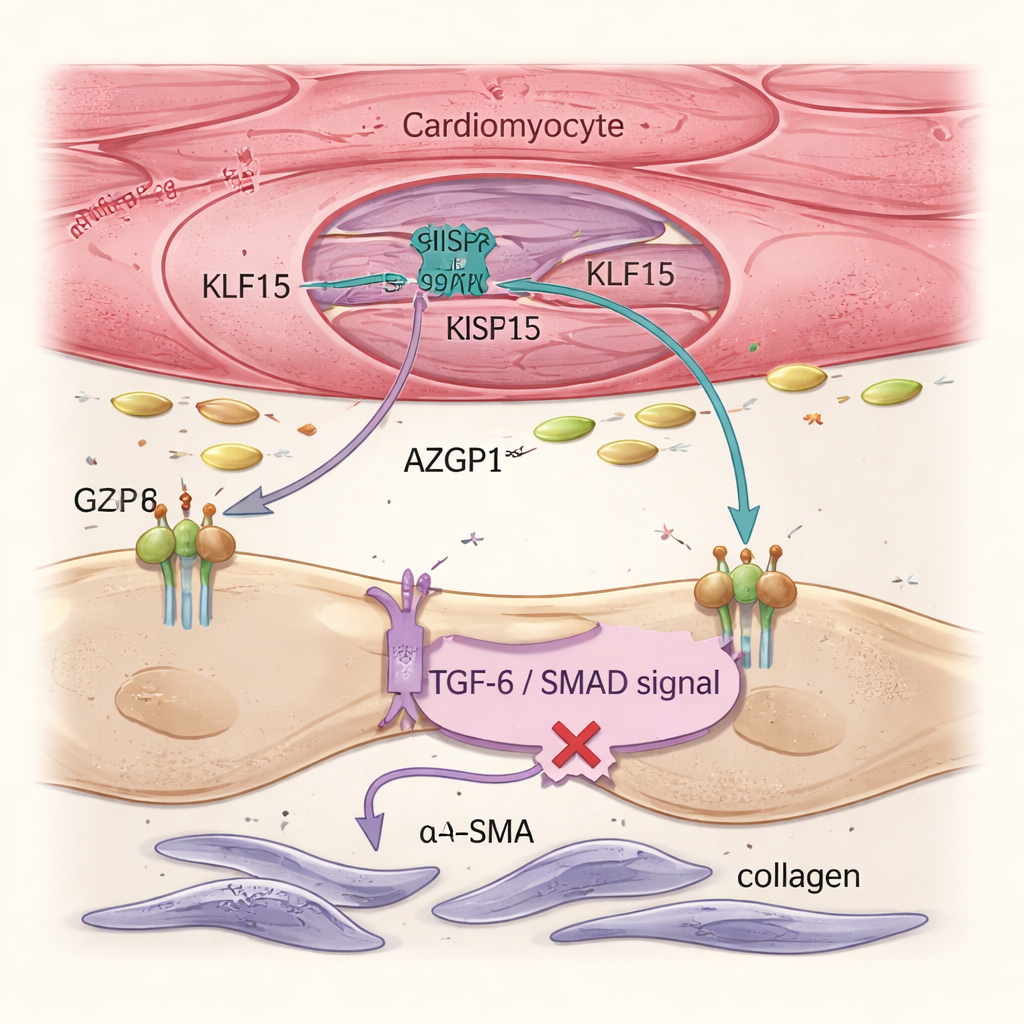
I modelli tissutali umani confermano il circuito protettivo
Per verificare se questi meccanismi fossero validi nelle cellule umane, i ricercatori hanno usato cardiomiociti derivati da cellule pluripotenti indotte coltivate in tessuti cardiaci ingegnerizzati tridimensionali. Sottoposti a carico meccanico che imita l’ipertensione, questi tessuti persero KLF15, attivarono geni legati allo stress e al programma fetale, si irrigidirono e la loro contrattilità diminuì — ricapitolando le caratteristiche della malattia. Il ripristino di KLF15 guidato da CRISPRa prevenne questo declino, preservò la generazione di forza e riportò l’espressione genica verso un profilo di metabolismo e struttura più maturi. Esperimenti dettagliati mostrarono che il TGF-β1, un noto segnale pro-fibrotico, riduce KLF15 nei cardiomiociti umani attraverso la via SMAD2/3, spiegando in parte come lo stress cronico porti a un rimodellamento maladattivo. Infine, il team ha costruito un sistema CRISPRa «mini» compatto basato su una variante di Cas9 più piccola che entra in un singolo vettore AAV9 ed è guidato da un promotore specifico per cardiomiociti. In fette di cuore umano in insufficienza tagliate con precisione, questo vettore aumentò con successo i livelli di KLF15 e migliorò le prestazioni contrattile nell’arco di giorni in coltura.
Un progetto per terapie geniche più delicate
Per un lettore non specialista, il messaggio centrale è che questo lavoro mostra come l’aumento mirato di un singolo regolatore protettivo all’interno delle cellule del muscolo cardiaco possa sia stabilizzare la loro identità sia inviare segnali che limitano la cicatrizzazione. Utilizzando un attivatore CRISPR che non taglia il DNA, l’approccio mette a punto i geni del cuore anziché inserire un gene artificiale. Lo studio definisce una via TGF-β → KLF15 → AZGP1 che collega lo stress meccanico al rimodellamento dannoso e dimostra, in topi, in modelli cellulari umani e in fette di tessuto cardiaco umano, che il ripristino di KLF15 può interrompere questa reazione a catena. Pur essendo ancora a livello preclinico, il sistema CRISPRa compatto e mirato ai cardiomiociti presentato qui offre una possibile roadmap per trattare forme comuni e non genetiche di scompenso cardiaco riprogrammando l’attività genica anziché riscrivere il genoma.
Citazione: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Parole chiave: scompenso cardiaco, KLF15, attivazione CRISPR, fibrosi cardiaca, AZGP1