Clear Sky Science · he
שונות בקצב ושגיאות רציפות בסדרות בפיטוגנטיקה בקנה מידה של מגפה
מדוע זה חשוב להתפרצויות עתידיות
כשוירוס חדש מתפשט ברחבי העולם, מדענים ממהרים לקרוא את הקוד הגנטי שלו ולשחזר את עץ המשפחה שלו. עצים אלה מסייעים לעקוב כיצד וריאנטים נוצרים, כמה מהר הם מתפשטים והאם אמצעי בקרה עובדים. אך במהלך COVID-19, מעבדות רצפו מיליוני גנומים של SARS‑CoV‑2 במהירות כה גבוהה שעיוותים מוסתרים ותקלות במידע התחילו לעוות את התמונה. מאמר זה מציג שיטות חדשות לניקוי ולפרשנות של מערכי נתונים גנטיים עצומים כאלה, ומציע תמונה ברורה יותר של האופן שבו וירוס מגפה אכן מתפתח ונע בקרב אוכלוסיות.
האתגר של הבנת מיליוני גנומים
אפידמיולוגיה גנומית הופכת גנומי וירוסים למידע פרקטי להחלטות בריאות הציבור. עבור SARS‑CoV‑2, שותפו יותר מ‑20 מיליון גנומים ברחבי העולם. הכלים האבולוציוניים המסורתיים נבנו לבעיות צנועות יותר, כגון השוואת גנים בין מינים, ולא לטיפול במיליוני רצפי וירוסים כמעט זהים שמגיעים בזמן אמת. בקנה מידה זה, שתי בעיות הופכות לבעייתיות במיוחד. ראשית, אתרים מסוימים בגנום הויראלי עוברים מוטציה בתדירות גבוהה יותר מאחרים, מה שעלול לגרום לוירוסים שאינם קרובים להיראות דומים באופן משונה. שנית, שגיאות טכניות חוזרות בריצוף ובעיבוד הנתונים יכולות לחקות מוטציות ממשיות. שני ההשפעות מייצרות "הדים שווא" בעץ האבולוציוני, ויוצרות אי‑ודאות לגבי אילו ענפים וקיבוצים ניתן לסמוך עליהם.
זיהוי אתרים המשתנים במהירות ושגיאות נסתרות
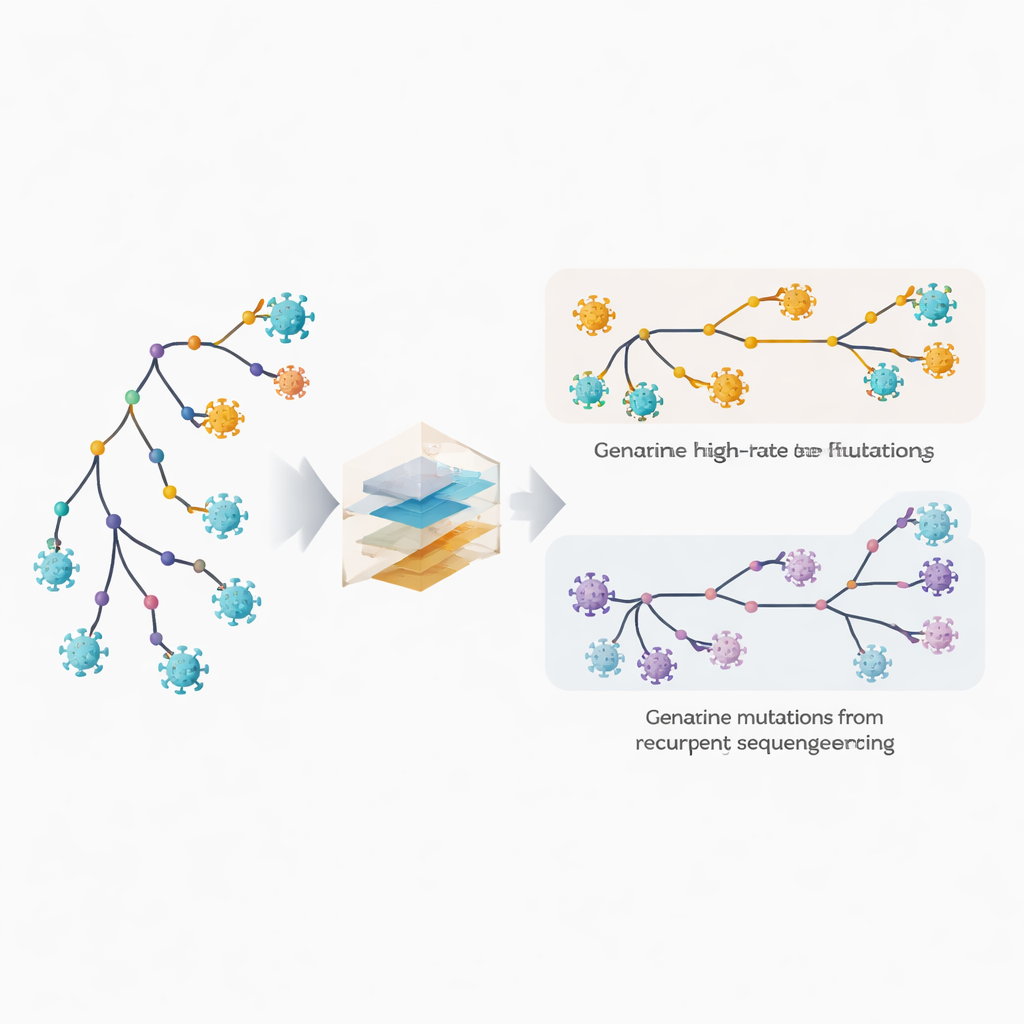
המחברים מרחיבים את תוכנת הפיטוגנטיקה שלהם, MAPLE, עם מודלים המתייחסים לכל מיקום בגנום הויראלי כאל בעל התנהגות נפרדת. במקום להניח מספר שיעורי מוטציה ממוצעים, השיטה מעריכה שיעור נפרד לכל אתר, ומנצלת את מספר הגנומים העצום הזמין. במקביל, היא מאפשרת לכל אתר להיות לו הסתברות משלו לשאת שגיאת ריצוף חוזרת או טעות בקביעת הקונצנזוס. הטריק המרכזי הוא להשוות כמה פעמים שינוי מופיע בענפים פנימיים עמוקים בעץ, המשקפים אירועים ישנים ומשותפים, לעומת הקצוות החיצוניים ביותר, שמתאימים לגנומים בודדים. מוטציות ביולוגיות אמיתיות נוטות להתחלק בין ענפים פנימיים לטרמינליים, בעוד ששגיאות טכניות מופיעות בעיקר בקצוות. על ידי ניצול תבנית זו, השיטה מסוגלת להפלות בין אבולוציה מהירה אמיתית לבין טעויות חוזרות.
אלגוריתמים מהירים יותר לעץ החיים הצפוף
טיפול במיליוני גנומים בדרך כלל ידרוש כוח חישוב עצום. כדי לשמור על ניתוח מעשי, הצוות עיצב מחדש את אופן שבו MAPLE מאחסן ומעדכן מידע על רצפים בעץ. במקום להשוות כל גנום לייחוס קבוע יחיד, התוכנה בוחרת "ייחוסים מקומיים" בתוך העץ ורושמת גנומים סמוכים כהבדלים יחסית לעוגנים אלה. ייצוג דחוס זה מזרז השוואות בין חלקים מרוחקים בעץ. שיפורים נוספים משכללים את אופן הוספת דגימות חדשות לעץ קיים, את כיוון התאמת אורך הענפים, ואיך בוחנים צורות עץ חלופיות סבירות, עם אפשרויות להריץ שלבים תובעניים ביותר במקביל על גבי מספר ליבות מעבד.
בדיקת השיטה וניקוי נתוני העולם האמיתי
כדי לבדוק שהמודלים שלהם פועלים, המחברים תחילה יצרו מערכי נתוני SARS‑CoV‑2 מדומים ריאליסטיים עם דפוסי מוטציה ידועים ושגיאות רצף מוטמעות. במבחנים אלה הגישה החדשה שחזרה עצים אבולוציוניים יותר נכונים וזיהתה שגיאות בודדות בדיוק גבוה, במיוחד כאשר נכללו עשרות אלפי גנומים או יותר. לאחר מכן פנו לנתונים אמיתיים, ניתחו מיליוני רצפי SARS‑CoV‑2 שלגביהם היו זמינים קריאות גולמיות. בהשוואת שני צינורות בניית קונצנזוס שונים, הם זיהו מיקומי גנום מסוימים שנפגעו שוב ושוב על ידי ארטיפקטים, כגון בעיות קשירת פריימר או קריאה המוטה על ידי הייחוס. אתרים חשודים אלה הוסתרו מניתוח נוסף, וגנומים שהראו סימני זיהום או דלקת מעורבת סוננו החוצה, מה שהניב יישור מפרושר של למעלה משני מיליון רצפים באיכות גבוהה.
תמונה עולמית ברורה יותר של עץ משפחת הוירוס
באמצעות מערך הנתונים המטוהר, המחברים שיחזרו עץ פיטוגנטי עולמי של SARS‑CoV‑2 ומיפו כיצד וריאנטים עיקריים קשורים זה לזה. העץ שלהם מציע לפעמים יחסים עדינים השונים מעצים ציבוריים קודמים, לעיתים בדרכים שדורשות פחות אירועי מוטציה ומתאימות טוב יותר למודל הסטטיסטי. המסגרת גם מדגישה מקומות שבהם תוויות קווים עשויות להיות לא עקביות עם ההיסטוריה הגנטית הבסיסית, ומסמנת אפשריים רקומביננטים או גנומים בעייתיים לבחינה מעמיקה יותר. אף שקיימים עדיין אתגרים — כגון התאמה-יתר כאשר הנתונים דלים, או השפעת דגימות מזוהמות בצורה חמורה — העבודה מראה כי כעת ניתן לבנות עצים אבולוציוניים בקנה מידה של מגפה שאמינים יותר. לציבור הרחב, המסקנה העיקרית היא שטיפול טוב יותר בשגיאות ובנקודות חמות למוטציה מוביל לתובנה חדה יותר לגבי האופן שבו פתוגנים מתפשטים ומשתנים, ועוזר למדענים ולרשויות הבריאות להגיב מהר ובביטחון רב יותר בהתפרצויות עתידיות.
ציטוט: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
מילות מפתח: גנומיקה של SARS-CoV-2, שיטות פיטוגנטיות, שגיאות בריצוף, שונות בקצב המוטציה, אפידמיולוגיה גנומית