Clear Sky Science · he
אימונותרפיה אנטי-TLR2 משנה העברה של α-סינוקלאין מנוירון לאוליגודנדרוציט במודלים של עכבר ובני אדם
מדוע המחקר הזה חשוב
תסמונת מערכות מרובות (MSA) היא הפרעה מוחית נדירה אך מתפתחת במהירות, שמשלבת תסמיני תנועה דמויי פרקינסון עם כשלי שיווי משקל ותפקוד אוטונומי, כגון נפילות בלחץ הדם. רופאים יכולים להקל על התסמינים, אך כרגע אינם יכולים לעצור את התקדמות המחלה. במחקר זה חושפים כיצד חלבון המקופל בצורה לא תקינה מתפשט בין תאי המוח ופוגע ב"בידוד החיווט" של המוח, ומראים שטיפול ממוקד בנוגדן יכול לקטוע תהליך זה במודלים של תאים ובעלי חיים. הממצא מצביע על אסטרטגיה דמוית-תרופה שמאוחר יותר עשויה לשנות את מהלך ה-MSA ולא רק לטפל בתסמיניו.
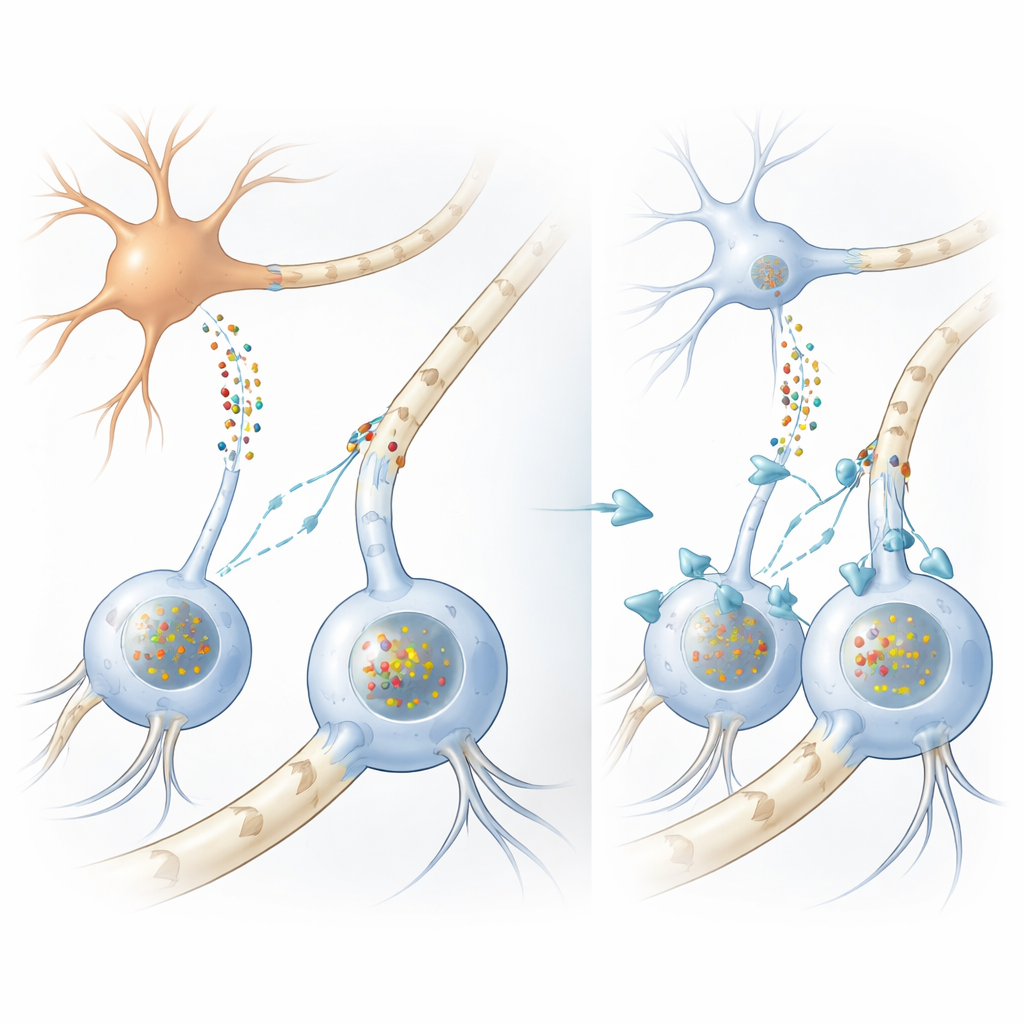
איך החיווט המוחי משתבש
ברבים מההפרעות התנועתיות, כולל מחלת פרקינסון, מצטברות בתוך תאי העצב צברי חלבון בשם אלפא-סינוקלאין. ב-MSA, עם זאת, הצברים הבולטים ביותר נוצרים בתוך תאי תמיכה הנקראים אוליגודנדרוציטים, שבדרך כלל עוטפים סיבי עצב במעטפות מיאלין שומניות המאיצות את האותות החשמליים. באופן בלתי צפוי, אוליגודנדרוציטים מייצרים מעט מאוד אלפא-סינוקלאין בעצמם, מה שמשאיר חידה ותיקה: מה מקור ההצטברויות הענקיות בחלבון בתאים הללו? החוקרים אישרו תחילה, בעזרת דגימות מוח אנושיות וניתוחים נרחבים של RNA, שאוליגודנדרוציטים אכן מייצרים הרבה פחות אלפא-סינוקלאין מאשר נוירונים, ומחזקים את הרעיון שהחלבון המזיק מגיע מלמטה החוצה.
חלבון שעובר מנוירון לתא תמיכה
כדי לבדוק זאת, הצוות בנה מספר מודלים משלימים. במקערות רקמה הם גידלו תאים דמויי-אוליגודנדרוציטים ממח עצביים אנושיים והחשיפו אותם לנוזל שנאסף מתאים דמויי-נוירון שתוכננו לשחרר כמויות גדולות של אלפא-סינוקלאין. תאי התמיכה לקחו על עצמם חלבון זה ופיתחו צברים הדומים מאוד להכללות הגליאליות הנצפות במוחות של חולי MSA, כולל התגים הכימיים ועוזרי החלבון המתאימים. כשבדקו זן עכבר המייצר אלפא-סינוקלאין אנושי מוטנט רק בנוירונים, הם שוב מצאו צברים של החלבון האנושי בתוך אוליגודנדרוציטים בחומר הלבן, אף על פי שתאים אלה לא הביעו את הגן האנושי. ביחד, הניסויים האלה מראים שאלפא-סינוקלאין יכול לנוע מנוירונים לאוליגודנדרוציטים וליצור בהם הכללות הדומות למחלה.
השער על פני תא
בהמשך, החוקרים שאלו כיצד החלבון נכנס לתוך האוליגודנדרוציטים. מחקרים קודמים זיהו את חיישן החיסון Toll-like receptor 2 (TLR2) על פני התא כאתר עגינה לאלפא-סינוקלאין בנוירונים ובמיקרוגליה. בחיפושי מערכי ביטוי גנים ממוחות של חולי MSA, הצוות מצא שאוליגודנדרוציטים בחולים נשאו רמות גבוהות מהרגיל של TLR2 בהשוואה לביקורות, וכי רמות TLR2 גבוהות נקשרו לרמות נמוכות יותר של גנים הקשורים למיאלין כמו חלבון המיאלין הבסיסי. קשר זה לא הופיע בכמה מערכי נתונים עצמאיים של פרקינסון, מה שמעיד על כך שרגישות האוליגודנדרוציטים לאלפא-סינוקלאין דרך TLR2 עשויה להיות תכונה מובחנת של MSA ולא מאפיין כללי של כל ההפרעות בסינוקלאין.
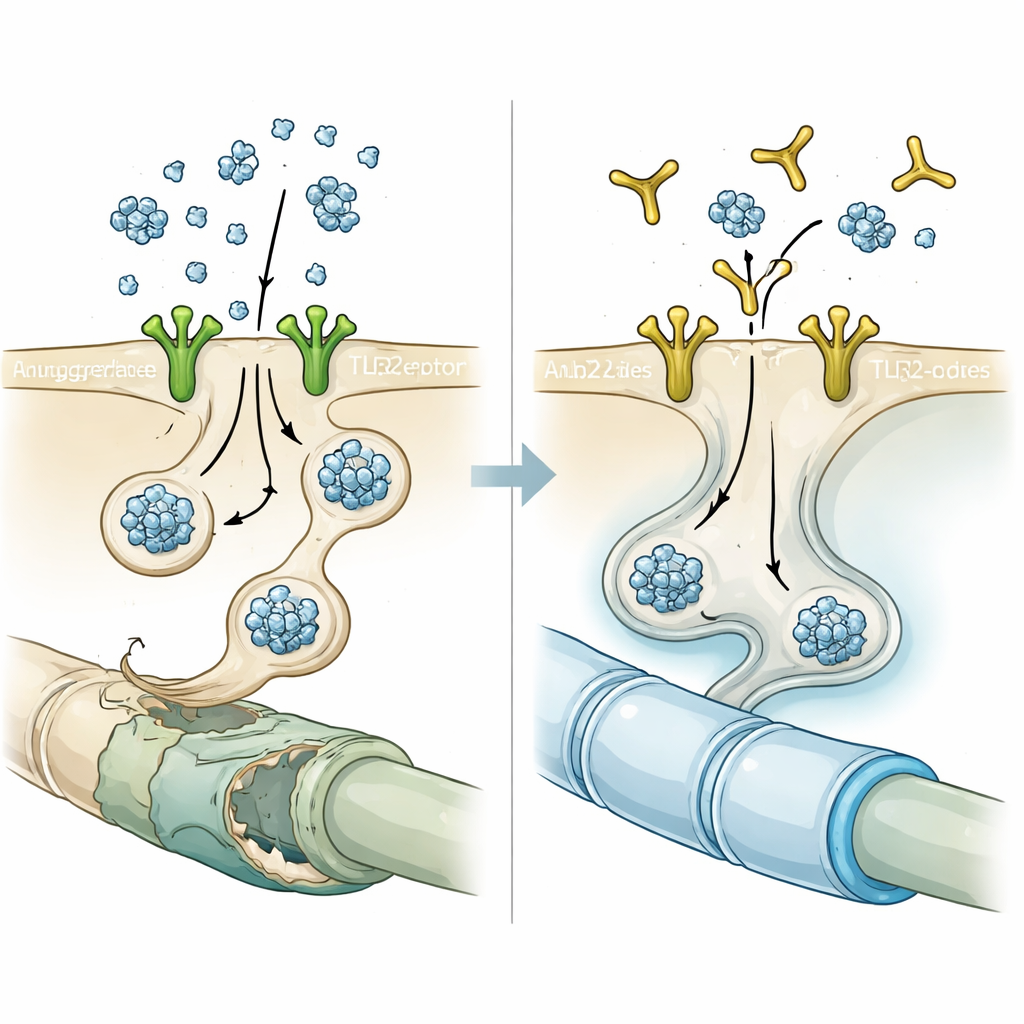
חסימת השער בעזרת נוגדן
בהסתמך על הרמז הזה, החוקרים בחנו את NM-101, נוגדן שתוכנן לקשור את TLR2 ולמנוע את הפעלתו. בתרביות תאים, טיפול מקדים קצר של אוליגודנדרוציטים ב-NM-101 לפני הוספת אלפא-סינוקלאין שמקורו בנוירונים הקטין באופן חד את מספר ועוצמת הצברים הדמויי-הכללה. בעכברים שהפרישו יתר של אלפא-סינוקלאין נוירונלי או שקיבלו הזרקות של סיבים של אלפא-סינוקלאין מוכנים מראש, הזנות שבועיות של NM-101 הפחיתו חלבון מצבר בחומר הלבן, הרגיעו תגובות דלקתיות של מיקרוגליה ואסטרוציטים, והקטינו את ההפעלה של אנזים דלקתי הנקרא קספאז-1 בתוך אוליגודנדרוציטים. בעלי החיים שטופלו חיו זמן ארוך יותר והופיעו טוב יותר במבחני תנועה, מה שמרמז שההשפעות המגנות של הנוגדן היו בעלות משמעות תפקודית, ולא רק סקרנות מיקרוסקופית.
הצלת הבידוד הפגוע
מכיוון שאוליגודנדרוציטים הם יוצרי המיאלין במוח, הצוות בדק האם העברת אלפא-סינוקלאין פגעה במיאלין והאם חסימת TLR2 יכלה לעזור. רצף RNA בתא יחיד של אוליגודנדרוציטים שמקורם בבני אדם וחשופים לאלפא-סינוקלאין מתנאי-נוירון גילה תמורות רחבות: ירידה במצב בוגר מייצר-מיאלין והיסט-עבר לפרופיל יותר נאיבי ודמוי-פרוגניטור, עם דיכוי של רבים מהגנים המרכזיים למיאלין. מחקרי ביטוי גנים מקבילים של אוליגודנדרוציטים שבודדו בלייזר ממטופלי MSA ומהמודל העכברי הראו חתימה משותפת: הפחתת ביטוי גנים המעורבים ביצירה ובתחזוקה של מיאלין. תחת המיקרוסקופ האלקטרוני, חומר לבן בעכברים עם אלפא-סינוקלאין הראה מעטפות מיאלין דקות ומבולגנות. טיפול ב-NM-101 הפך חלק מהשינויים האלה, העבה את המיאלין, השיב רמות של חלבוני מיאלין ונירמל את הביטוי של גנים הנדרשים לבשלות האוליגודנדרוציטים.
מה המשמעות לכל זה עבור טיפולים עתידיים
המחקר תומך בסיפור ברור: ב-MSA, אלפא-סינוקלאין המיוצר על ידי נוירונים יכול להתפשט לאוליגודנדרוציטים דרך TLR2 על פני השטח שלהם, שם הוא מצטבר, מעורר דלקת, מסטה את תוכנית ההתפתחות של התאים ומסיג את שכבת המיאלין של חיווט המוח. על ידי חסימת TLR2 בעזרת נוגדן ממוקד, החוקרים הצליחו לקטוע את שרשרת האירועים הזו בעכברים ובמודלים של תאים אנושיים, ולהפחית הכללות רעילות, להרגיע דלקת, לתקן מיאלין ולשפר הישרדות ותנועה. אמנם NM-101 עצמו עדיין זקוק לניסויים קפדניים באדם, המחקר מקבע את ההעברה התלוית-TLR2 של החלבון כמניע מרכזי של פתולוגיה דמוית-MSA ומבליט את האימונותרפיה נגד TLR2 כאסטרטגיה מבטיחה להאט או למנוע את המחלה ההרסנית הזו.
ציטוט: Bae, EJ., Ham, S., Jeong, Y.W. et al. Anti-TLR2 immunotherapy modulates neuron-to-oligodendrocyte propagation of α-synuclein in mouse and human models. Nat Commun 17, 2175 (2026). https://doi.org/10.1038/s41467-026-68870-x
מילות מפתח: תסמונת מערכות מרובות, אלפא-סינוקלאין, אוליגודנדרוציטים, נזק למיאלין, אימונותרפיה