Clear Sky Science · fr
Génération moléculaire 3D contrainte par interactions utilisant un modèle de diffusion permet la modélisation pharmacophorique basée sur la structure pour la conception de médicaments
Pourquoi concevoir de meilleurs médicaments est si difficile
La découverte moderne de médicaments repose souvent sur la capacité à faire entrer une petite molécule dans une protéine comme une clé dans une serrure. Mais la clé doit faire plus que s’emboîter : elle doit établir le bon ensemble de petites attractions — telles que de faibles forces électriques directionnelles et des zones évitant l’eau — afin que le médicament reste lié de façon forte et spécifique. L’univers chimique est astronomiquement vaste, bien au‑delà de ce que contiennent aujourd’hui les bases de données, si bien que les chercheurs cherchent des moyens plus intelligents d’inventer de nouvelles clés à partir de zéro tout en préservant ces schémas de contact cruciaux.
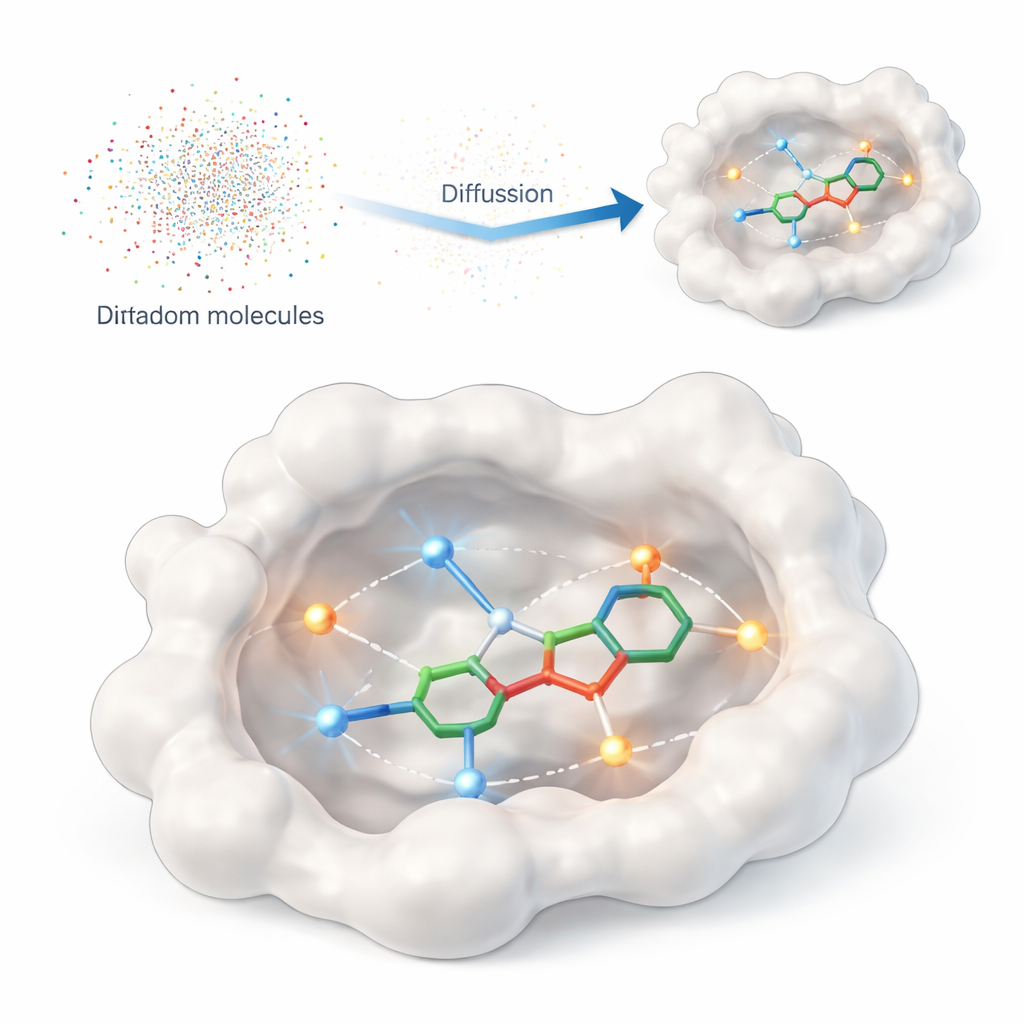
Apprendre à l’ordinateur ce qui compte vraiment
Cette étude présente DiffPharma, un cadre computationnel qui génère des molécules 3D de type médicament directement à l’intérieur du site de liaison d’une protéine. Plutôt que de demander à l’algorithme de parcourir d’énormes catalogues de composés existants, DiffPharma crée de nouvelles molécules atome par atome, guidé par la manière dont elles doivent interagir avec la protéine. La méthode repose sur une classe moderne de modèles génératifs appelés modèles de diffusion, qui partent d’un bruit aléatoire et « débruitent » progressivement ce bruit pour obtenir un objet structuré — dans ce cas, une molécule 3D nichée dans la poche protéique.
Coder la poignée de main de la protéine
Pour indiquer au modèle ce qui importe à la surface de la protéine, les auteurs représentent les contacts clés par de petites « particules d’interaction » dispersées le long des trajets entre la protéine et une molécule de référence. Deux types d’interactions courants sont mis en avant : les liaisons hydrogène, qui agissent comme des aimants directionnels entre atomes spécifiques, et les contacts hydrophobes, où des régions « grasses » se regroupent loin de l’eau. Des réseaux neuronaux distincts apprennent la géométrie et la chimie de chaque type d’interaction, ainsi que la forme globale de la poche de liaison, puis une architecture de fusion spéciale combine ces points de vue en une image unique et cohérente qui guide la génération des molécules.
Dans quelle mesure reproduit‑il les véritables schémas de liaison ?
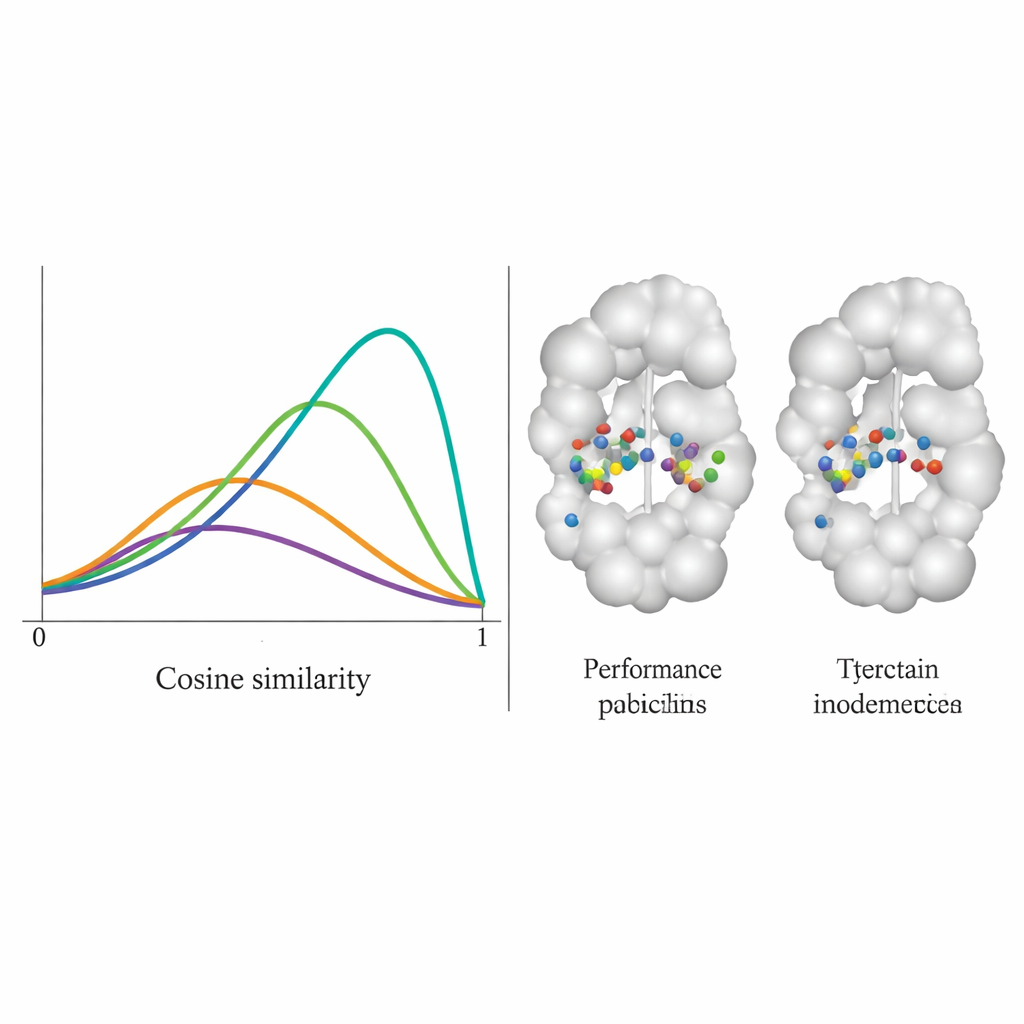
L’équipe a testé DiffPharma sur 100 paires protéine–molécule différentes et a évalué dans quelle mesure les nouvelles molécules reproduisaient fidèlement les schémas de contact originaux, résidu par résidu. Ils ont mesuré cela à l’aide d’un score de similarité cosinus entre 0 et 1, où 1 signifie un accord parfait. La distribution des scores pour DiffPharma culminait autour de 0,9, ce qui signifie qu’en moyenne les mêmes résidus protéiques formaient les mêmes types d’interactions clés que dans les structures de référence — bien mieux que six méthodes concurrentes. Fait important, le modèle accomplissait cela tout en produisant une variété de formes moléculaires, et les composés générés conservaient des longueurs de liaison, des angles et une géométrie 3D globaux réalistes, typiques de molécules réelles et stables.
De la théorie à des candidats médicamenteux pratiques
Au‑delà des benchmarks, les auteurs ont demandé si DiffPharma pouvait concevoir des candidats médicaments plausibles pour des cibles réelles. Pour deux enzymes bien étudiées — la kinase AKT et une β‑lactamase liée à la résistance aux antibiotiques — la méthode a généré des molécules qui préservaient les patrons d’interaction essentiels des ligands connus tout en utilisant souvent des échafaudages chimiques différents, une forme souhaitable de « scaffold hopping » en chimie médicinale. Dans une étude de cas plus exigeante sur la protéase principale du SARS‑CoV‑2, DiffPharma a été orienté par des choix d’interactions spécifiques puis examiné avec des simulations de dynamique moléculaire et des estimations d’énergie de liaison. Les molécules générées sous des contraintes d’interaction plus strictes formaient des complexes plus stables et présentaient parfois des énergies de liaison prédictives plus favorables qu’un inhibiteur de référence connu. Notamment, le système a même redécouvert ce composé de référence — bien qu’il n’apparaisse jamais dans l’entraînement — uniquement à partir de la structure protéique et des instructions d’interaction.
Ce que cela signifie pour les médicaments de demain
Pour un non‑spécialiste, DiffPharma peut être vu comme un outil de conception intelligent et conscient de la 3D pour les molécules médicamenteuses : donné la forme d’une poche protéique et un schéma souhaité de « poignées de main », il propose des clés chimiquement raisonnables qui s’emboîtent et interagissent de la bonne manière. Bien qu’il n’optimise pas encore toutes les propriétés qu’un médicament requiert, comme la solubilité ou le métabolisme, la méthode préserve de manière fiable la carte de contacts cruciale à la surface de la protéine et explore de nouvelles régions de l’espace chimique au‑delà des catalogues actuels. Cette approche guidée par les interactions peut aider les chercheurs à passer plus rapidement des données structurales sur des protéines liées à une maladie à des points de départ expérimentaux diversifiés et réalistes pour le développement de médicaments.
Citation: Sako, M., Yasuo, N. & Sekijima, M. Interaction-constrained 3D molecular generation using a diffusion model enables structure-based pharmacophore modeling for drug design. npj Drug Discov. 3, 8 (2026). https://doi.org/10.1038/s44386-026-00040-x
Mots-clés: conception de médicaments basée sur la structure, modèles génératifs moléculaires, modélisation pharmacophorique, interactions protéine–ligand, protéase principale du SARS‑CoV‑2