Clear Sky Science · fr
Docking compétitif guidé par l’IA pour le criblage virtuel et la prédiction de l’efficacité des composés
Recherches plus intelligentes pour de nouveaux médicaments
Trouver de nouveaux médicaments ressemble un peu à chercher une aiguille dans une botte de foin composée de millions de molécules. Cette étude montre comment les progrès récents de l’intelligence artificielle peuvent accélérer et réduire le coût de cette recherche en aidant les scientifiques à prédire quelles molécules sont le plus susceptibles d’adhérer à une protéine liée à une maladie et de fonctionner effectivement comme médicaments. Plutôt que de tester chimiquement une substance à la fois au laboratoire, les auteurs utilisent des modèles d’IA pour organiser des concours virtuels entre molécules et laisser les gagnants remonter en haut de la liste.
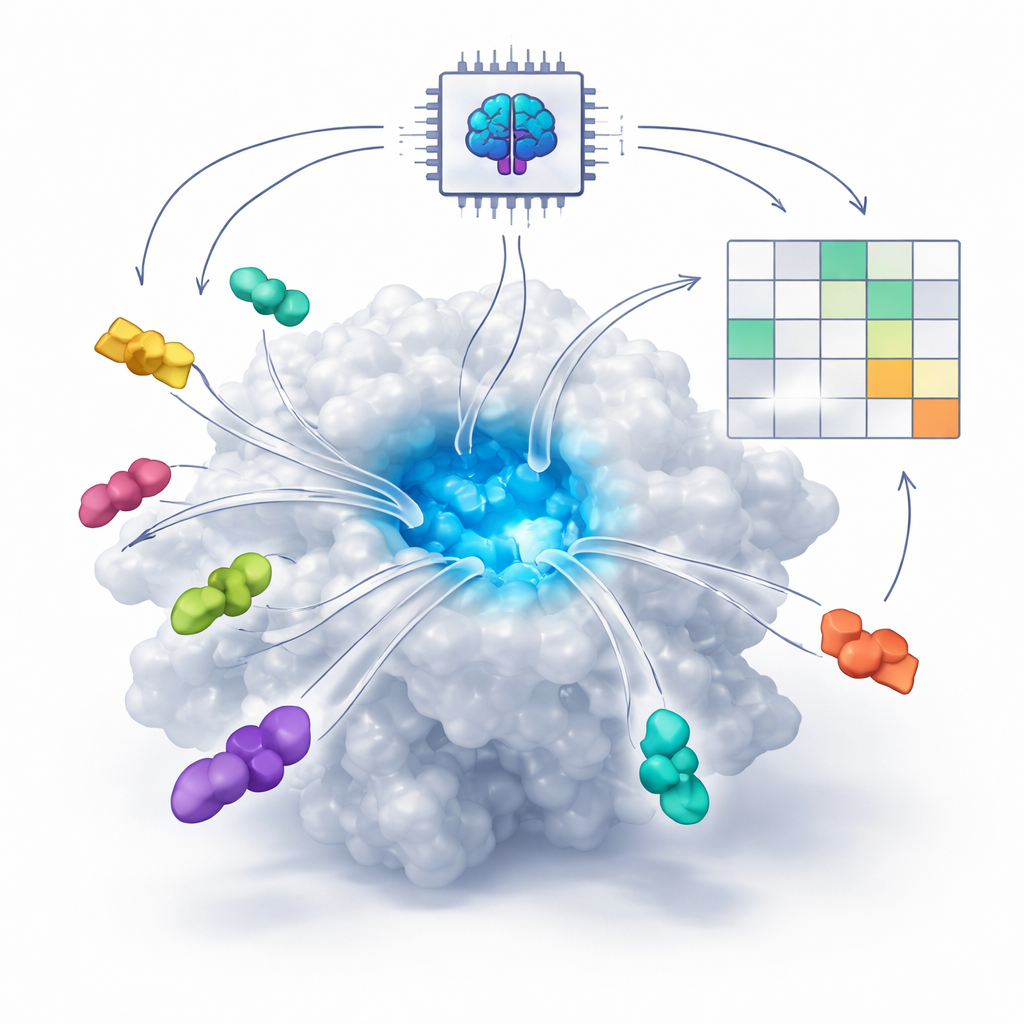
Comment l’IA apprend à voir les ajustements clé-serrure moléculaires
Beaucoup de médicaments modernes agissent en s’insérant dans de petites poches des protéines, comme une clé dans une serrure. Traditionnellement, des programmes informatiques tentaient de prédire cet ajustement à l’aide d’équations de physique estimant les forces entre atomes. Ces dernières années, cependant, de nouveaux systèmes d’IA basés sur la diffusion et la co-repliement — tels qu’AlphaFold3 et Boltz — ont appris à partir d’un grand nombre de structures protéine–molécule connues. Ces systèmes peuvent désormais « imaginer » comment une protéine et un médicament potentiel pourraient se replier ensemble en trois dimensions, même lorsqu’aucune structure expérimentale n’existe. La question centrale abordée par les auteurs est de savoir si ces outils d’IA peuvent faire plus que dessiner des images plausibles — peuvent-ils aussi distinguer les bons médicaments des mauvais ?
Vrais ligands vs. imposteurs
L’équipe a d’abord testé 16 protéines bien étudiées ainsi qu’une enzyme bactérienne plus complexe appelée gyrase de l’ADN. Pour chaque protéine, ils ont demandé aux modèles d’IA de placer à la fois des inhibiteurs actifs connus et un ensemble de molécules « hors cible » non apparentées dans le même site de liaison. Plutôt que de se fier à une seule prédiction, ils ont examiné la constance avec laquelle l’IA positionnait chaque molécule sur de nombreuses exécutions. Les vrais inhibiteurs avaient tendance à revenir au même emplacement et à la même orientation à plusieurs reprises, se regroupant à quelques trillionièmes de mètre les uns des autres. Les molécules inactives vagabondaient davantage et se trouvaient souvent plus éloignées de la poche. Cette idée simple — la convergence de pose — s’est avérée être un signal fort indiquant qu’un composé s’adapte réellement à sa cible protéique.
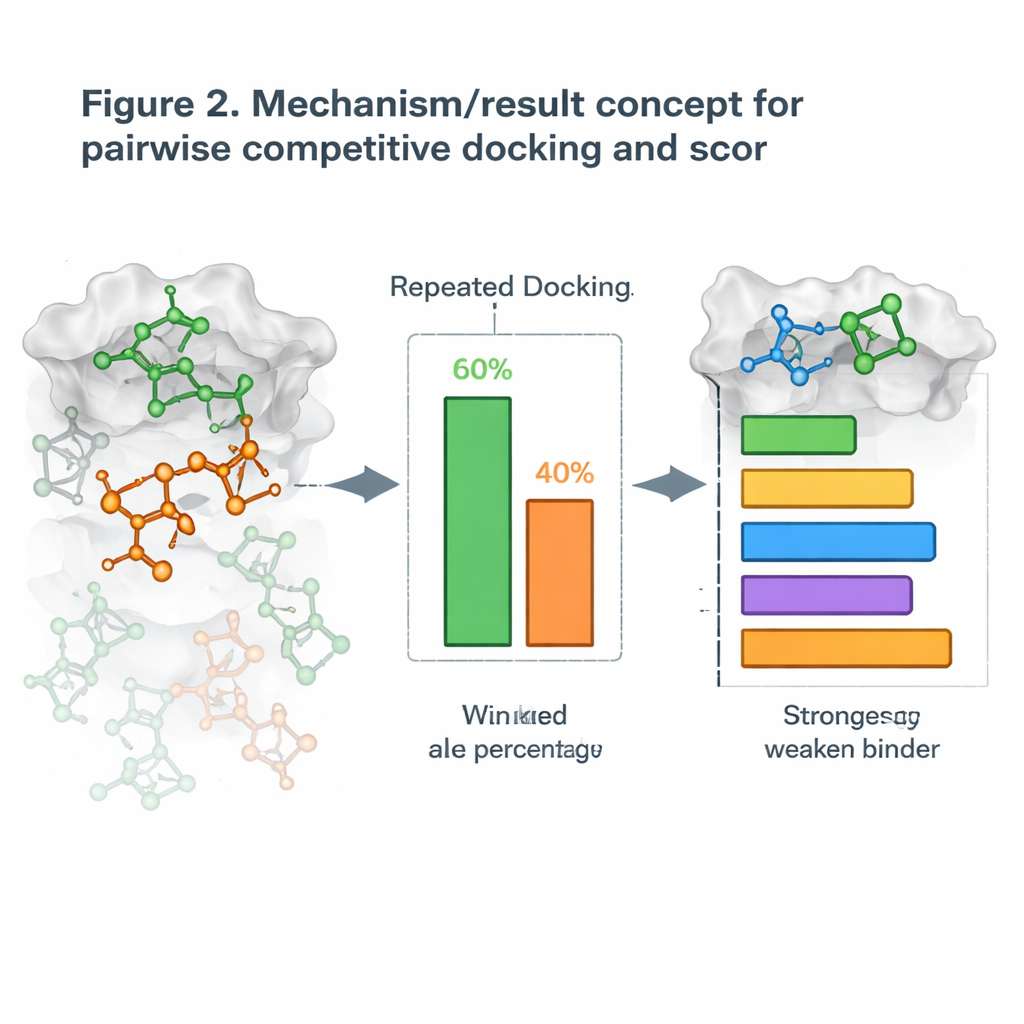
Transformer le docking en compétition directe
En s’appuyant sur cela, les auteurs ont introduit une nouvelle stratégie qu’ils appellent docking compétitif par paires. Plutôt que de docker une molécule à la fois, ils en dockent deux candidates simultanément avec la protéine et les laissent « se disputer » la même poche. Après de nombreuses répétitions, la molécule qui occupe le site le plus souvent est déclarée gagnante de ce duel. En exécutant toutes les paires possibles, ils construisent une table de victoires-défaites et calculent un Score de Docking Compétitif pour chaque molécule, un peu comme classer des joueurs dans un tournoi à la ronde. Lorsque ces scores ont été comparés aux mesures expérimentales réelles de l’affinité d’inhibition des molécules, les classements concordaient souvent bien, certains systèmes protéiques montrant un accord quasi parfait.
Du criblage virtuel à la conception de meilleurs antibiotiques
La gyrase de l’ADN, une enzyme essentielle pour les bactéries, a servi de cas test détaillé. Cette protéine possède plusieurs poches ciblées par différentes classes d’antibiotiques, y compris les fluoroquinolones largement utilisées. Les modèles d’IA pouvaient généralement placer chaque classe de médicaments dans sa poche correcte, et les scores de docking compétitif reflétaient approximativement leurs puissances mesurées. Les auteurs ont ensuite étendu leur approche à un criblage virtuel de plus de 3 000 médicaments approuvés, demandant quelles molécules compétiient le mieux pour le site des fluoroquinolones. Leur stratégie en deux étapes — utiliser d’abord une compétition « tous ensemble » pour sélectionner des candidats probables, puis filtrer selon la condensation de leurs positions dans la poche — a fortement enrichi la fraction de véritables fluoroquinolones tout en écartant les candidats plus faibles. Enfin, ils ont utilisé un générateur de molécules piloté par l’IA pour proposer de nouvelles structures de type fluoroquinolone et appliqué le docking compétitif pour trouver une poignée de composés avec une affinité prédite meilleure et des propriétés drug-like acceptables.
Promesses, limites et implications pour les patients
L’étude montre que les modèles d’IA modernes peuvent faire plus que produire des structures protéine–médicament plausibles : lorsqu’ils sont utilisés dans un cadre compétitif, ils peuvent aider à classer des composés d’une manière qui reflète souvent les données expérimentales réelles. Cela ne remplace pas le travail en laboratoire — les performances dépendent fortement de la protéine considérée, certaines poches sont mal prédictives, et les modèles d’IA peuvent échouer pour des molécules très grandes ou atypiques. Mais à mesure que ces modèles et leurs données d’entraînement s’améliorent, des approches comme le docking compétitif par paires pourraient rendre la découverte précoce de médicaments beaucoup plus efficace. Pour les patients, cela pourrait éventuellement se traduire par un développement plus rapide de médicaments ciblés, y compris de nouveaux antibiotiques capables de suivre le rythme des bactéries résistantes.
Citation: Mirgaux, M., Barcelli, V., Chua, A.C.Y. et al. AI-guided competitive docking for virtual screening and compound efficacy prediction. npj Drug Discov. 3, 6 (2026). https://doi.org/10.1038/s44386-026-00039-4
Mots-clés: découverte de médicaments par IA, criblage virtuel, docking moléculaire, liaison protéine-ligand, conception d’antibiotiques