Clear Sky Science · fr
Calcul scientifique économe en énergie grâce à des réservoirs chimiques
Pourquoi transformer la chimie en calcul a de l’importance
Les supercalculateurs modernes consomment d’énormes quantités d’électricité pour simuler le climat, concevoir de nouveaux médicaments ou entraîner l’intelligence artificielle. À mesure que nous approchons des limites physiques des puces traditionnelles, il devient plus difficile et plus coûteux d’extraire davantage de performance par watt. Cet article explore une voie radicalement différente : utiliser de vraies réactions chimiques comme moteur du calcul scientifique. En traitant les molécules et leurs interactions comme les pièces mobiles d’un ordinateur, les auteurs exposent comment de futures machines pourraient résoudre des équations complexes avec beaucoup moins d’énergie que le matériel numérique actuel.
Des cellules vivantes aux calculateurs chimiques
Les cellules vivantes sont des virtuoses de la résolution de problèmes. Elles gèrent en permanence des milliers de réactions pour s’adapter, croître et survivre, tout en utilisant remarquablement peu d’énergie. Au cœur de ce comportement se trouvent les réseaux de réactions chimiques : des réactions interconnectées dont les vitesses et les concentrations évoluent dans le temps. Ces réseaux peuvent être décrits par des équations différentielles ordinaires, le même langage mathématique utilisé pour modéliser tout, des épidémies aux écoulements turbulents. L’idée centrale de ce travail est que si la chimie suit déjà ces équations, on pourrait l’exploiter directement pour effectuer les calculs que les scientifiques réalisent aujourd’hui sur silicium.
Comment des équations deviennent des réseaux de réactions
Les auteurs présentent ChemComp, un cadre logiciel qui prend un système d’équations différentielles et le convertit systématiquement en un réseau abstrait de réactions. ChemComp utilise des technologies de compilation modernes pour décomposer un problème mathématique en motifs pouvant être représentés par des réactions idéalisées, puis les organise en un réseau avec des espèces, des connexions et des constantes de vitesse bien définies. Ces réactions abstraites ne correspondent pas encore à de vraies molécules, mais elles forment le plan d’un ordinateur chimique. Le cadre peut ensuite rechercher dans des bases de données biochimiques des motifs réactionnels réels qui se comportent de façon similaire, en favorisant des options pratiques, sûres et potentiellement économes en énergie dans un cadre expérimental.
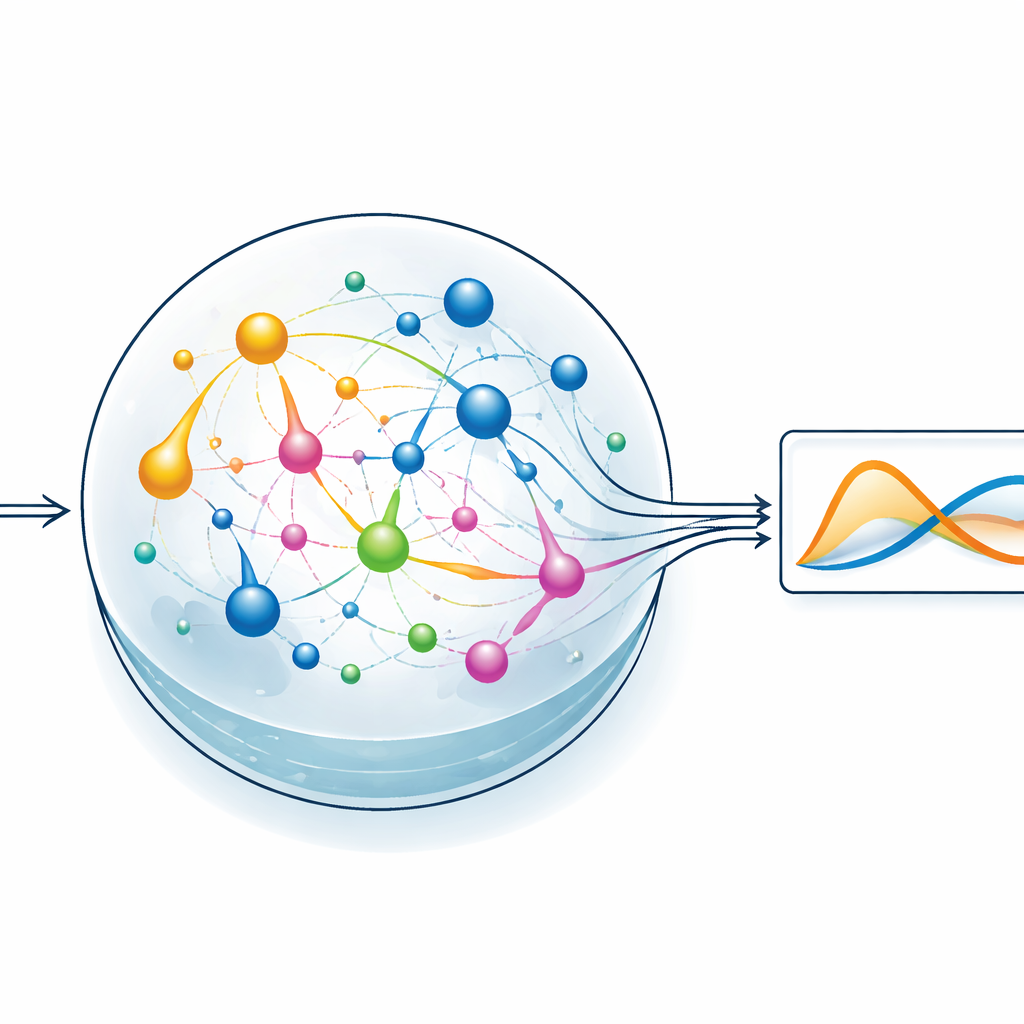
Laisser un réservoir chimique faire le travail difficile
Pour tester l’idée, l’équipe se concentre sur un style d’apprentissage automatique appelé reservoir computing. Ici, un système dynamique fixe transforme un signal d’entrée en un motif interne riche et complexe, et seule une couche de lecture simple est entraînée pour produire la sortie désirée. Dans la version de ChemComp, le réservoir est un ensemble de réactions dans un récipient bien agité ; les variations de concentrations chimiques forment les états internes. Les auteurs compilent un système classique à deux variables connu sous le nom de modèle Sel’kov–Schnakenberg — initialement utilisé pour étudier des oscillations métaboliques — en réseaux de réactions candidats. Ils simulent ensuite la réponse de ces réseaux au fil du temps lorsqu’ils sont pilotés par des flux de produits entrant et sortant du récipient, et utilisent une régression linéaire élémentaire pour combiner les traces de concentration en une approximation de la solution cible.
Tester des réseaux chimiques simples et plus riches
Les chercheurs comparent deux réservoirs candidats : l’un avec seulement deux espèces chimiques et deux réactions, l’autre avec cinq espèces et cinq réactions. Les deux réseaux reçoivent des concentrations initiales et des débits adaptés, puis sont simulés en fonctionnement. Même le système le plus petit peut reproduire grossièrement le comportement oscillant des équations cibles, mais le réseau plus grand fait sensiblement mieux, réduisant l’erreur tant pendant l’entraînement que lors des tests. En balayant différentes concentrations initiales et constantes de vitesse, les auteurs cartographient les régions où le système chimique correspond le mieux à la dynamique souhaitée. Chaque réaction joue en pratique le rôle d’une fonction de base dans un problème d’ajustement de courbe : plus les réactions disponibles sont variées, plus il est facile d’approcher un comportement complexe, au prix d’une complexité accrue du système.
Vers un calcul peu énergivore en laboratoire
Au-delà des simulations, l’article se tourne vers des dispositifs pratiques. Il examine comment le choix des réactions doit équilibrer consommation d’énergie, contrôlabilité par des enzymes ou catalyseurs, et capacité à mesurer des espèces clés en temps réel, par exemple par des méthodes optiques ou électrochimiques. Les auteurs suggèrent que des plateformes microfluidiques futures pourraient héberger des réseaux réactionnels soigneusement choisis, avec un contrôle spatial des entrées et des capteurs intégrés. Bien que de nombreux défis d’ingénierie subsistent — depuis la mise en correspondance des équations avec la chimie réelle jusqu’à la gestion du bruit et des limites de mesure — l’étude montre que des systèmes réactionnels modestes peuvent déjà émuler les solutions d’équations différentielles couplées. Pour un lecteur non spécialiste, le message central est que la chimie elle‑même peut agir comme un ordinateur analogique, ouvrant la voie à des calculs scientifiques qui s’appuient sur les processus économes en énergie que la nature a perfectionnés pendant des milliards d’années.
Citation: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Mots-clés: informatique chimique, informatique économe en énergie, computing par réservoir, réseaux de réactions chimiques, équations différentielles ordinaires