Clear Sky Science · fr
Un cadre de bout en bout pour la réactivité en catalyse hétérogène
Pourquoi accélérer la conception des catalyseurs est important
La société moderne dépend des catalyseurs pour fabriquer carburants, plastiques, engrais et d’innombrables produits du quotidien. Pourtant, trouver de meilleurs catalyseurs revient souvent à chercher une aiguille dans une botte de foin, car chaque matériau peut favoriser des milliers de réactions microscopiques possibles simultanément. Cet article présente CARE, un nouveau cadre computationnel qui utilise des règles intelligentes et l’apprentissage automatique pour cartographier et simuler ces toiles réactionnelles emmêlées beaucoup plus rapidement et de façon plus complète qu’auparavant. Ce faisant, il promet d’orienter des technologies énergétiques plus propres et des procédés chimiques plus efficaces tout en réduisant considérablement les coûts de calcul.
Démêler des voies réactionnelles encombrées
À la surface d’un catalyseur solide, les molécules entrantes ne suivent pas simplement une unique trajectoire nette du réactif au produit. Elles traversent plutôt un labyrinthe d’intermédiaires transitoires et de voies concurrentes. Les méthodes informatiques traditionnelles s’appuient sur l’intuition humaine pour sélectionner un ensemble limité d’étapes possibles puis utilisent des calculs quantiques pour évaluer leurs énergies. Cela fonctionne pour de petits réseaux mais s’effondre rapidement à mesure que les systèmes se complexifient, négligeant des voies rares qui peuvent gouverner l’activité à long terme, la désactivation ou la sélectivité. CARE relève ce défi en construisant automatiquement de très grands réseaux réactionnels à partir de règles simples de construction, garantissant que tous les événements plausibles de rupture et de formation de liaisons entre carbone, hydrogène et oxygène sont inclus, même ceux que les chimistes pourraient normalement écarter.
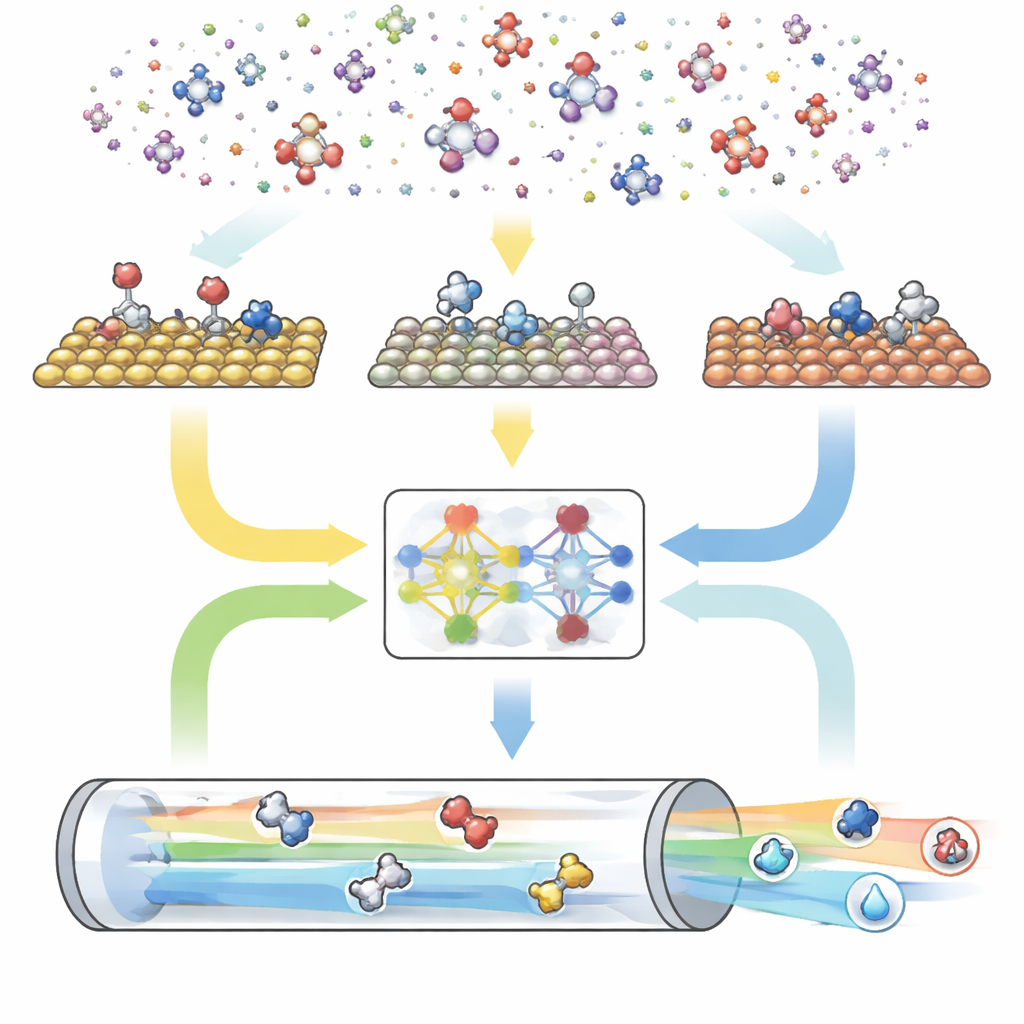
Un moteur numérique en trois parties pour les réactions
CARE est conçu comme une chaîne de traitement de bout en bout comportant trois modules principaux. D’abord, un générateur basé sur des règles définit « l’espace chimique » en choisissant le nombre maximal d’atomes de carbone et d’oxygène puis en appliquant des modèles simples pour créer toutes les molécules correspondantes et leurs formes liées à la surface. Ensuite, un module d’évaluation énergétique fait appel à des modèles modernes d’apprentissage automatique — en particulier un réseau de neurones graphes nommé GAME-Net-UQ — pour estimer les énergies des intermédiaires et des états de transition sur de nombreux plans métalliques. Ce modèle considère chaque structure comme un réseau d’atomes et de liaisons, fournit à la fois une énergie et une incertitude, et atteint une précision de l’ordre de quelques dixièmes d’électron-volt tout en restant léger et rapide. Enfin, un solveur microcinétique utilise ces énergies pour calculer comment l’ensemble des réactions se déroule sous des conditions réalistes de température, pression, tension et pH, en prédisant les vitesses de réaction globales, les couvertures de surface et la sélectivité des produits.
Tests réels : molécules combustibles et chimie climatique
Pour montrer que CARE n’est pas qu’un exercice théorique, les auteurs l’appliquent à trois problèmes industriels d’importance croissante. Pour la décomposition du méthanol — une réaction importante pour le stockage de l’hydrogène — ils génèrent un réseau modeste et l’évaluent sur de nombreux catalyseurs métalliques et surfaces cristallines. CARE reproduit la tendance en « volcan » familière de l’activité et identifie correctement le ruthénium comme l’un des meilleurs performeurs, en accord avec les expériences, mais en une fraction infime du temps de calcul nécessaire pour des calculs quantiques complets. Ensuite, ils passent à la conversion électrochimique du dioxyde de carbone sur cuivre, en se concentrant sur la formation de produits à trois atomes de carbone tels que le 1-propanol et le propylène. En incluant des étapes spécifiques tenant compte des protons, des électrons et des conditions de solution, CARE capture comment le pH et la tension appliquée déplacent les voies et prédit correctement que le 1-propanol est favorisé par rapport au propylène, faisant écho à des études détaillées antérieures.
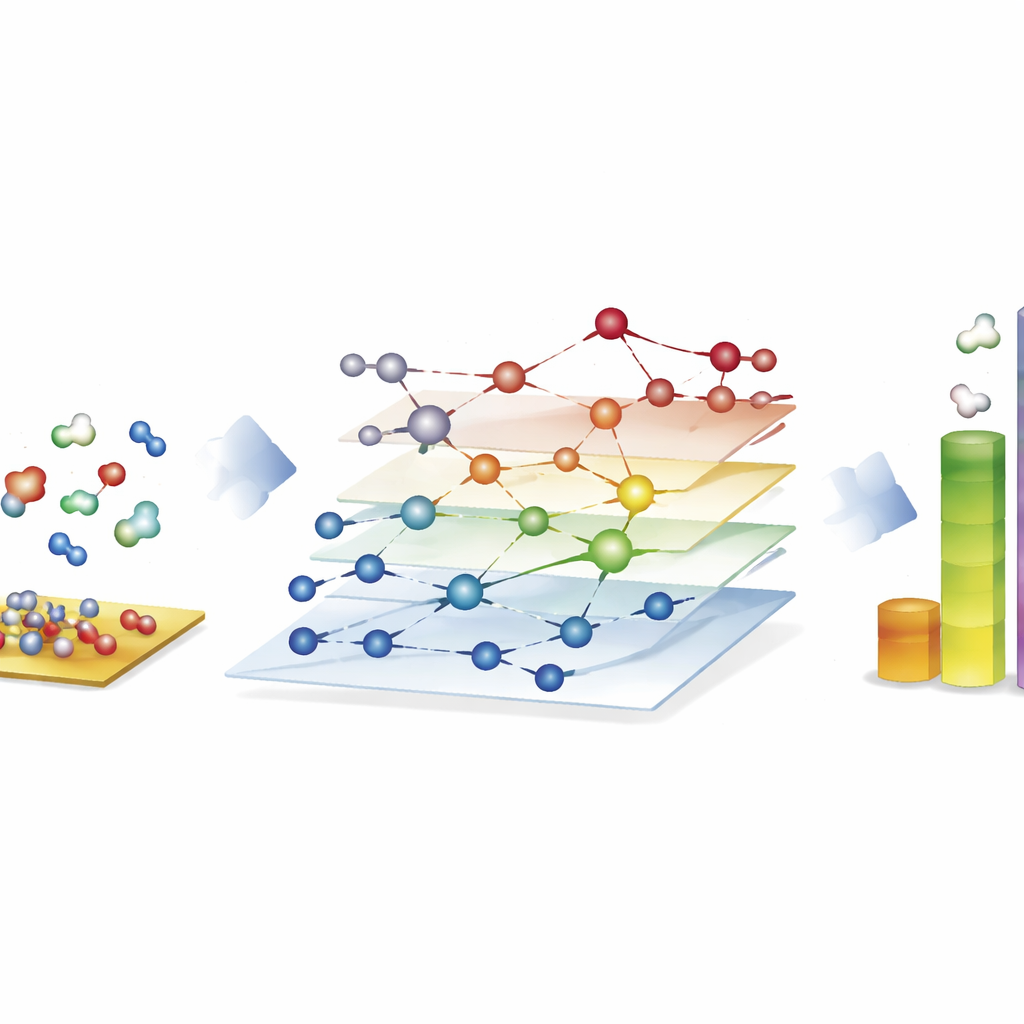
Explorer d’énormes toiles réactionnelles pour les carburants synthétiques
La démonstration la plus frappante provient du procédé de Fischer–Tropsch, qui transforme des mélanges de monoxyde de carbone et d’hydrogène en hydrocarbures à chaîne longue pour carburants et produits chimiques. Ici, les auteurs construisent des réseaux comprenant près de 40 000 espèces de surface et environ 370 000 réactions élémentaires — bien au-delà de ce que les études traditionnelles basées sur la mécanique quantique peuvent explorer complètement. Avec CARE, ils évaluent tous les intermédiaires et les barrières réactionnelles clés sur des surfaces de cobalt, fer, nickel et ruthénium en seulement quelques heures sur du matériel standard, un gain de vitesse d’environ un million de fois comparé aux calculs quantiques directs. Les simulations microcinétiques sur ces réseaux reproduisent des tendances connues : le cobalt et le fer forment préférentiellement des chaînes d’hydrocarbures plus longues, le fer produit davantage de dioxyde de carbone via des réactions secondaires, et le nickel tend vers une hydrogénation plus prononcée. Bien que certains détails, comme les rendements en méthane, restent imparfaits, le cadre révèle quelles étapes de formation de liaisons dominent la croissance de chaîne et souligne où les modèles nécessitent encore des améliorations.
Ce que cela signifie pour les catalyseurs de demain
Pour les non-spécialistes, le message clé est que CARE offre une manière pratique d’explorer d’énormes espaces réactionnels à la surface de catalyseurs qui étaient auparavant hors de portée. En automatisant la génération de réseaux, en intégrant des modèles substituts d’apprentissage automatique rapides pour les énergies quantiques et en résolvant efficacement la cinétique résultante, il peut classer des catalyseurs candidats, identifier des conditions opératoires prometteuses et découvrir des voies inattendues avec beaucoup moins de biais humain et de coûts de calcul. Si les auteurs soulignent des défis restants — tels que la meilleure prise en compte des surfaces encombrées, des effets de solvant et des réseaux encore plus vastes — le travail ouvre la voie à un futur où les ordinateurs peuvent rapidement cribler des réactions complexes, de la réduction du dioxyde de carbone au recyclage des plastiques et à la valorisation de la biomasse, en guidant les expériences vers les idées les plus prometteuses plutôt qu’en laissant la découverte au tâtonnement.
Citation: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Mots-clés: catalyse hétérogène, réseaux réactionnels, apprentissage automatique, modélisation microcinétique, synthèse de Fischer–Tropsch