Clear Sky Science · fr
Un cadre génomique complet pour identifier les gènes prédisposant aux cancers du sein ou de l’ovaire déficients en réparation par recombinaison homologue
Pourquoi certaines familles présentent un risque accru de cancer
De nombreuses femmes atteintes d’un cancer du sein ou de l’ovaire ont des antécédents familiaux marqués, et pourtant les tests génétiques n’apportent souvent pas d’explication claire. Ce déficit, parfois qualifié « d’héritabilité manquante », laisse les familles sans réponses et peut limiter l’accès à des dépistages ou des traitements adaptés, comme les médicaments ciblés. Cette étude visait à bâtir une nouvelle méthode plus puissante pour chercher dans notre ADN des gènes de risque cachés en combinant plusieurs types d’informations génomiques et cliniques plutôt qu’en les explorant isolément. 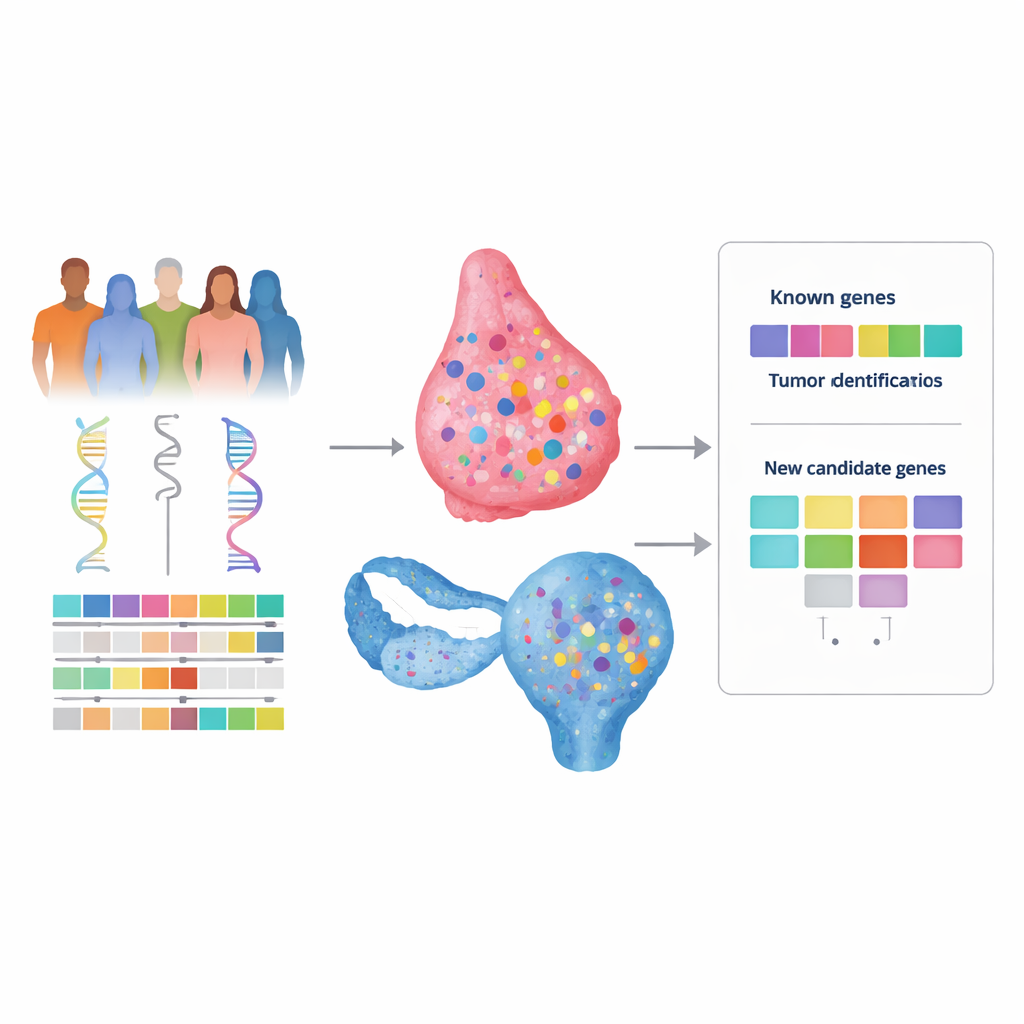
Rechercher l’empreinte d’un défaut de réparation de l’ADN
Les chercheurs se sont concentrés sur un type particulier de réparation de l’ADN, la réparation par recombinaison homologue, qui corrige normalement les cassures dangereuses du matériel génétique. Lorsque ce système de réparation fait défaut, les cellules accumulent une « signature mutationnelle » identifiable — un motif caractéristique de variations dans l’ADN tumoral qui agit comme une empreinte moléculaire. Ce motif est particulièrement fréquent dans certains cancers du sein et de l’ovaire difficiles à traiter. L’équipe a émis l’hypothèse que si une tumeur présente cette empreinte, elle a probablement perdu les deux copies fonctionnelles d’un gène de réparation de l’ADN, connu ou non, et que cela pourrait remonter à des variations héritées dans ce gène.
Construire un cadre combinant génétique et clinique
Pour tester cette idée, les auteurs ont analysé l’ADN de centaines de patientes atteintes de cancers du sein et de l’ovaire issues du Cancer Genome Atlas, qui fournit à la fois des données sanguines (héréditaires) et tumorales (acquises), ainsi que des informations cliniques. Ils ont exploré l’exome entier — la portion codante du génome — à la recherche de variantes héritées rares susceptibles d’être délétères et présentant aussi un « second coup » dans la tumeur, comme la perte de la copie saine restante. Pour chaque gène, ils ont ensuite évalué si de tels événements en deux temps étaient plus fréquents dans les tumeurs portant la signature de défaut de réparation que dans celles qui en étaient dépourvues. De façon cruciale, ils ne se sont pas limités à un panel prédéfini de gènes, ce qui a permis l’émergence de candidats inattendus.
Vérifier la méthode et identifier de nouveaux suspects
Comme validation, le cadre a correctement mis en évidence les gènes bien connus BRCA1 et BRCA2 comme fortement associés à la signature de défaut de réparation dans les deux types de cancer, confirmant que l’approche fonctionne comme prévu. Dans le cancer du sein, il a aussi signalé un gène supplémentaire, THBS4, et suggéré des rôles possibles pour KIF13B et TESPA1. Toutefois, un examen détaillé au cas par cas a montré que les altérations de THBS4 apparaissaient souvent en conjonction avec d’autres événements déjà établis liés à la réparation, ce qui en fait un candidat moins convaincant. 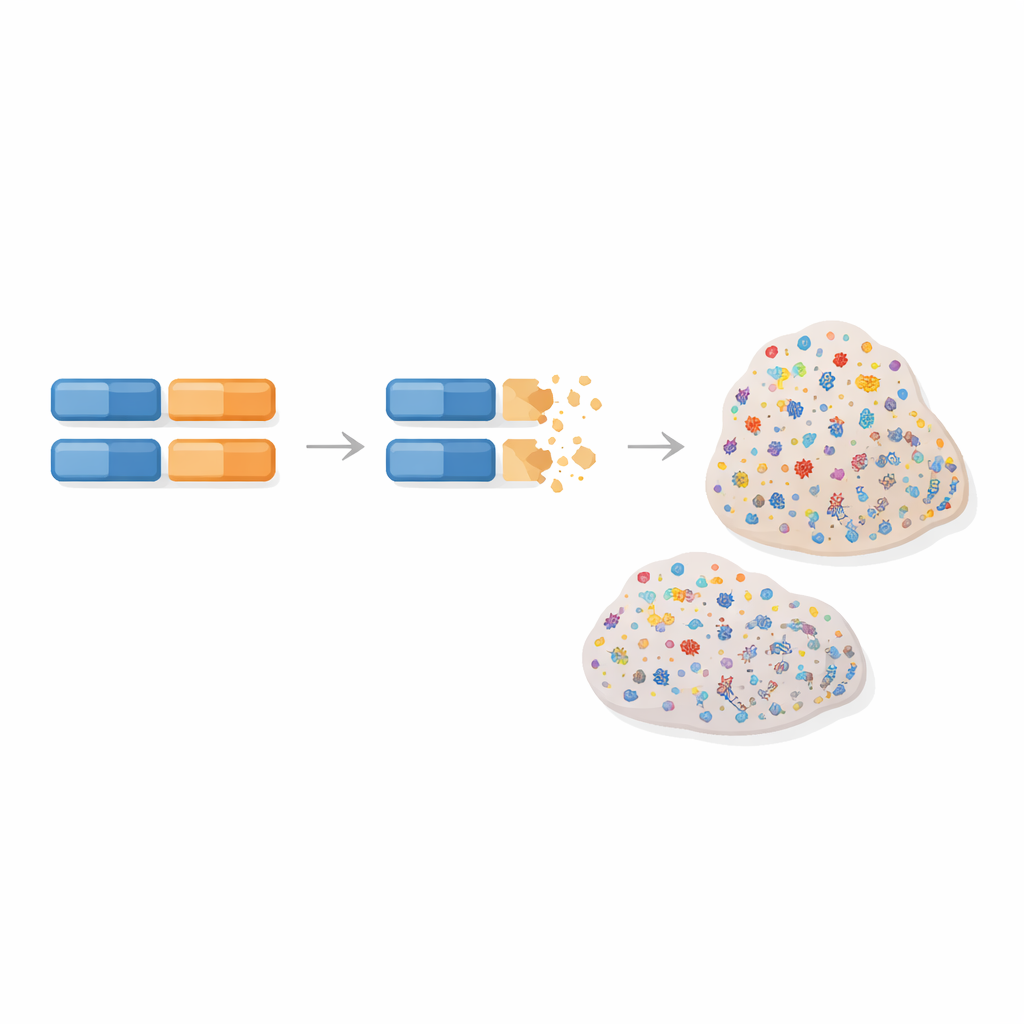
Se focaliser sur les tumeurs à haut risque inexpliquées
Pour dépasser l’analyse statistique, les chercheurs ont intégré des détails cliniques tels que le sous‑type tumoral, l’âge au moment du diagnostic et l’ascendance. Ils se sont concentrés sur les patientes dont les tumeurs présentaient nettement la signature de défaut de réparation et appartenaient à des groupes cliniques déjà associés à cette biologie — les cancers du sein de type basal et les cancers séreux de haut grade de l’ovaire — mais qui ne présentaient aucun événement de type BRCA connu. Chez ces patientes, ils ont recherché à nouveau des variantes héritées accompagnées d’un second coup, cette fois au sein d’une vaste liste sélectionnée de gènes du cancer et de réparation de l’ADN. Cette perspective « clinico‑génomique » a mis en lumière plusieurs gènes impliqués dans la réparation des cassures double brin de l’ADN et la voie étroitement liée de l’anémie de Fanconi, notamment RAD51B, RAD54B, RAD54L, FANCD2 et d’autres, comme contributeurs plausibles au risque héréditaire.
Implications pour les patientes et la recherche future
L’étude ne prétend pas avoir définitivement démontré de nouveaux gènes de risque : le nombre de patientes affectées par chaque gène candidat reste encore faible, et des cohortes plus larges et plus diversifiées seront nécessaires pour confirmer leur rôle. Les auteurs fournissent cependant une feuille de route réutilisable : une manière de combiner ADN hérité et tumoral, signatures mutationnelles caractéristiques et données cliniques pour prioriser systématiquement les gènes susceptibles d’expliquer des cancers familiaux inexpliqués. À terme, l’application de ce cadre à des jeux de données plus étendus et à d’autres types de cancer pourrait réduire l’écart d’« héritabilité manquante », affiner les panels de tests génétiques et aider davantage de patientes à comprendre leur risque personnel ainsi que leurs options de prévention et de traitements ciblés.
Citation: Camacho-Valenzuela, J., Matis, T., Roca, C. et al. A comprehensive genomic framework for identifying genes predisposing to homologous recombination repair-deficient breast or ovarian cancer. BJC Rep 4, 15 (2026). https://doi.org/10.1038/s44276-026-00218-w
Mots-clés: déficit de recombinaison homologue, génétique du cancer du sein, cancer de l’ovaire, gènes de réparation de l’ADN, susceptibilité au cancer