Clear Sky Science · fr
Expression hématopoïétique de cIAP2 entraîne inflammation et insuffisance cardiaque après infarctus du myocarde
Pourquoi calmer l’inflammation après un infarctus est important
Survivre à un infarctus n’est que le début de l’histoire. Dans les jours et semaines qui suivent, le système immunitaire se précipite pour nettoyer les tissus endommagés et amorcer la réparation. Mais si cette réponse inflammatoire est trop intense ou trop prolongée, elle peut transformer une guérison utile en lésion cardiaque durable et en insuffisance cardiaque. Cette étude met au jour un interrupteur moléculaire clé à l’intérieur des cellules immunitaires d’origine sanguine qui entretient ce feu inflammatoire — et montre qu’éteindre cet interrupteur peut protéger le cœur dans des modèles expérimentaux.
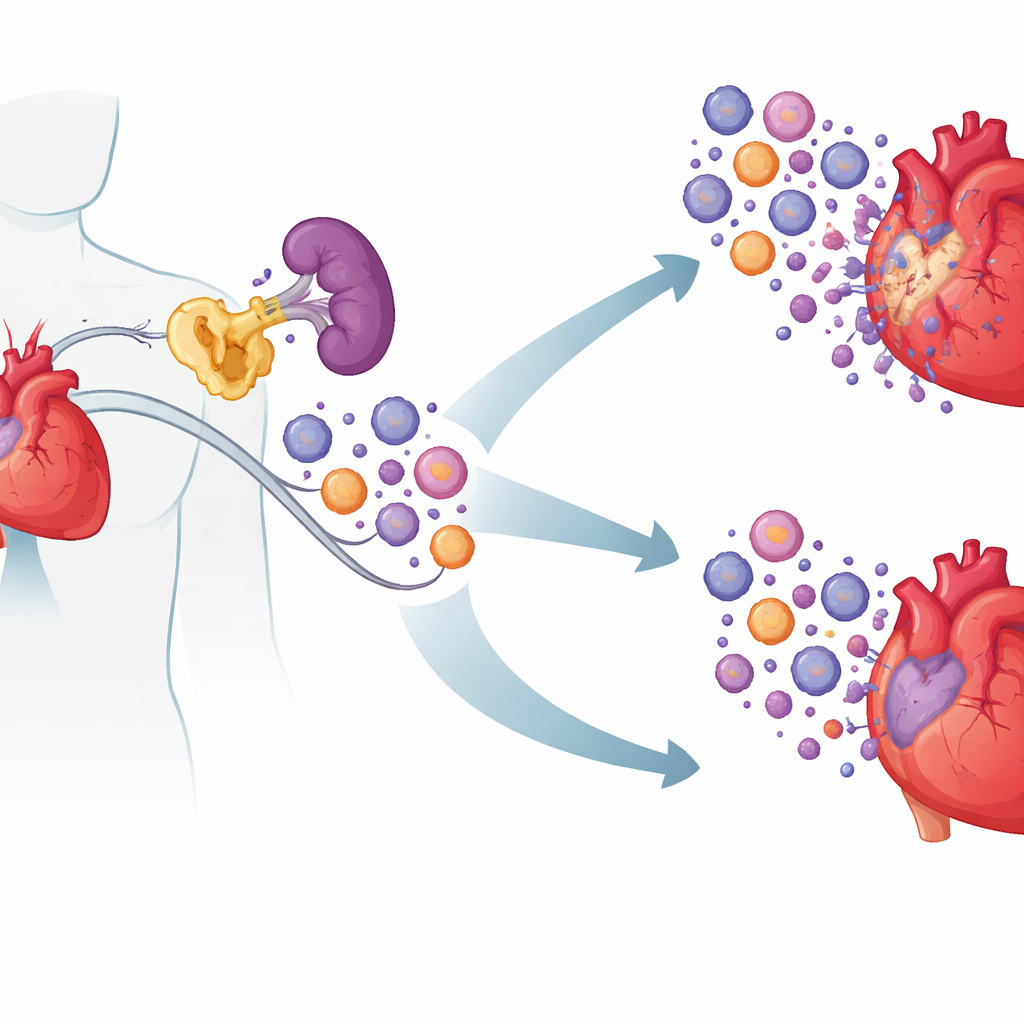
Un coupable caché à l’intérieur des cellules immunitaires
Les chercheurs se sont concentrés sur une protéine nommée cIAP2, mieux connue pour aider les cellules cancéreuses à éviter la mort. À partir d’échantillons sanguins de patients présentant des troubles cardiaques aigus, ils ont observé des niveaux de cIAP2 plus élevés chez les personnes ayant récemment fait un infarctus et chez celles souffrant d’insuffisance cardiaque d’origine ischémique que chez des individus sains ou des patients atteints de maladie coronarienne stable. Les tissus cardiaques humains et murins montraient le même schéma : cIAP2 montait en flèche peu après un infarctus, alors que sa proche homologue cIAP1 ne le faisait pas. En explorant des jeux de données d’expression génique existants, l’équipe a constaté que les niveaux de cIAP2 augmentaient de concert avec des gènes associés à des cellules inflammatoires de type myéloïde agressives, suggérant que cIAP2 pourrait amplifier la réponse immunitaire post-infarctus plutôt que de simplement refléter les lésions.
Moins de cIAP2, moins de dommages au cœur
Pour tester la causalité, l’équipe a comparé des souris normales à des souris génétiquement conçues pour être déficientes en cIAP2. Après un infarctus expérimental, les animaux dépourvus de cIAP2 présentaient des cicatrices plus petites, une fonction de pompage meilleure et moins d’accumulation de liquide dans les poumons, autant de signes d’un cœur en meilleure santé. Ces bénéfices apparaissaient chez les mâles comme chez les femelles. La microscopie a montré moins de cellules musculaires cardiaques mourantes dans les zones frontières critiques, et les analyses moléculaires ont révélé des niveaux plus faibles de marqueurs de stress et de remodelage des semaines plus tard. En revanche, l’inactivation de cIAP1 n’offrait pas la même protection et pouvait même aggraver les résultats dans certains cas, indiquant un rôle délétère et particulier pour cIAP2 dans ce contexte.
Le rôle de la rate comme réservoir inflammatoire
La clé était l’endroit où cIAP2 agissait. En échangeant la moelle osseuse entre souris normales et souris déficientes en cIAP2, les chercheurs ont montré que cIAP2 à l’intérieur des cellules hématopoïétiques (formatrices de sang) était responsable d’une grande partie des lésions. Lorsque les cellules immunitaires manquaient de cIAP2 mais que le reste de l’organisme était normal, le cœur était mieux protégé ; l’échange inverse aggravait les dommages. En se penchant sur les organes immunitaires, ils ont découvert qu’après un infarctus, la rate servait de réservoir produisant des cellules myéloïdes — neutrophiles, monocytes inflammatoires et cellules dendritiques — qui affluaient ensuite vers le cœur. Chez les souris sans cIAP2, ces cellules myéloïdes spléniques étaient moins nombreuses et plus susceptibles de mourir, tandis que les lymphocytes étaient en grande partie épargnés. Les signaux liés aux voies inflammatoires étaient atténués, ce qui suggère que cIAP2 aide normalement les cellules myéloïdes à survivre et à continuer de répondre aux signaux de danger.
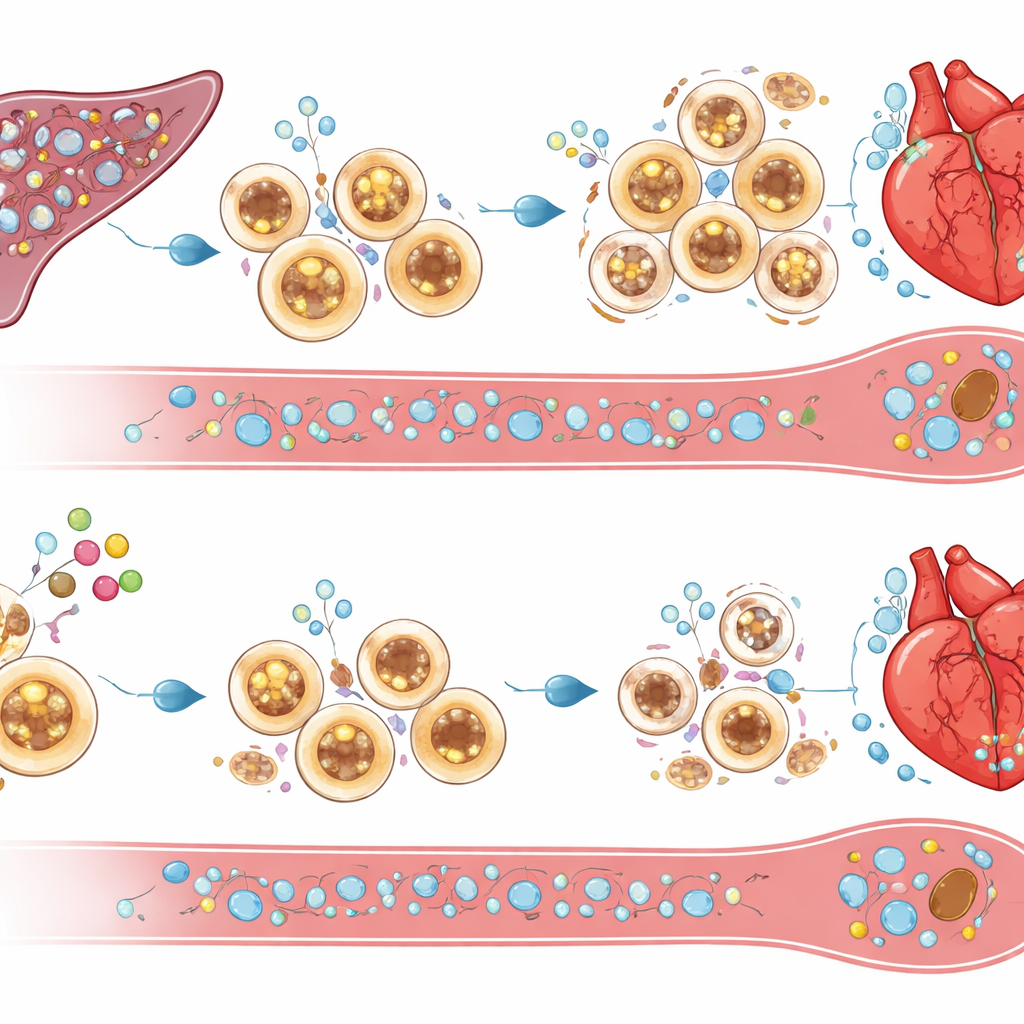
Transformer les signaux de survie en nettoyage auto-limitant
Qu’est-ce qui élimine l’excès de cellules inflammatoires lorsque cIAP2 est absent ? L’étude pointe vers des molécules inductrices de mort telles que TRAIL et son récepteur DR5, ainsi que des signaux liés au TNF, qui étaient sur-régulés dans la rate et la moelle osseuse des souris déficientes en cIAP2 après infarctus. Le blocage expérimental de TRAIL a permis de sauver les cellules spléniques de la mort, de rétablir une forte infiltration immunitaire du cœur et d’effacer les bénéfices fonctionnels liés à l’absence de cIAP2. Cela suggère que cIAP2 protège normalement les cellules myéloïdes de ces signaux de mort, leur permettant de s’accumuler et de prolonger l’inflammation. Sans cIAP2, ces mêmes signaux élaguent le réservoir splénique, réduisant l’apport de cellules agressives qui, autrement, envahiraient le cœur lésé.
Cibler cet interrupteur pour des thérapies futures
Fait important, l’équipe a montré que cette voie peut être ciblée par une classe existante de petites molécules appelées mimétiques de Smac, actuellement étudiées en cancérologie. En utilisant le composé LCL161, ils ont déclenché de manière sélective la dégradation des protéines cIAP dans les cellules immunitaires spléniques peu après un infarctus, sans éliminer les protéines protectrices dans le tissu cardiaque lui-même. Les souris traitées présentaient moins de cellules inflammatoires en circulation, des cicatrices plus petites, une meilleure fonction cardiaque et une survie améliorée par rapport aux animaux non traités. Une seule administration à faible dose, donnée un jour après l’infarctus, suffisait à induire une mort contrôlée des cellules myéloïdes spléniques, à augmenter localement les niveaux de TRAIL et à réduire l’inflammation cardiaque, tandis que le nombre total de cellules immunitaires était rétabli au bout de quatre semaines. Ensemble, ces résultats positionnent cIAP2 comme un facteur de survie central pour les cellules inflammatoires après une lésion cardiaque et suggèrent qu’une inhibition ciblée et transitoire de cIAP2 pourrait offrir une nouvelle approche de type immunothérapeutique pour prévenir l’insuffisance cardiaque après un infarctus.
Citation: Smyth, D., Zhang, L., Al-Khalaf, M. et al. Hematopoietic expression of cIAP2 drives inflammation and heart failure after myocardial infarction. Nat Cardiovasc Res 5, 246–261 (2026). https://doi.org/10.1038/s44161-026-00782-x
Mots-clés: infarctus du myocarde, inflammation, cellules immunitaires, insuffisance cardiaque, mimétique de Smac