Clear Sky Science · fr
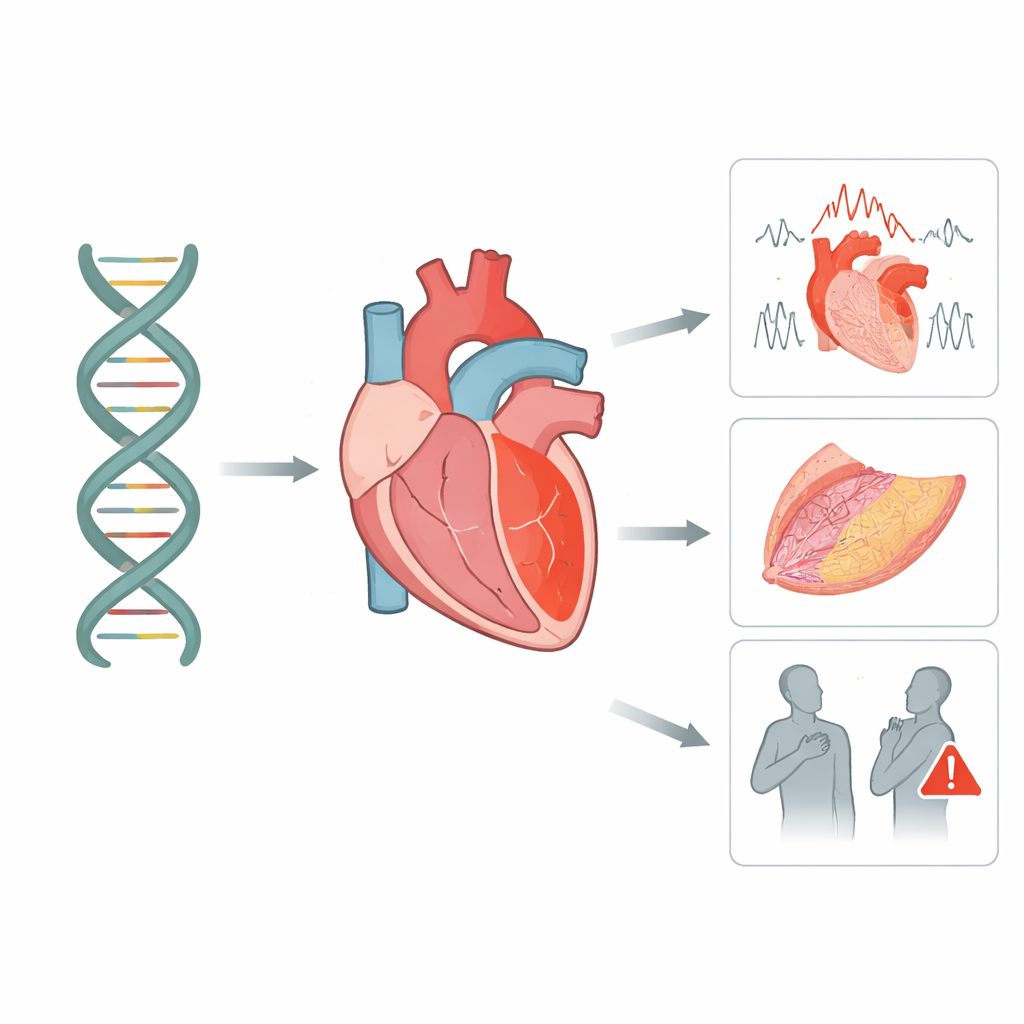
Évaluation intégrative génomique et bibliographique de la cardiomyopathie arythmogène liée à la desmoglycine‑2 avec validation sur une cohorte italienne
Pourquoi ce gène cardiaque compte pour les familles
De nombreux incidents cardiaques soudains chez des personnes jeunes et par ailleurs en bonne santé ne sont pas dus au hasard : ils sont, au moins en partie, écrits dans leur ADN. Cet article explore une protéine « colle » clé du cœur appelée desmogleine‑2 et montre comment de petites altérations de son gène peuvent affaiblir le muscle cardiaque, perturber son rythme électrique et augmenter le risque d’événements dangereux. En combinant de grands bases de données génétiques et un groupe de patients italiens soigneusement suivi, les chercheurs fournissent des réponses plus nettes pour les familles qui se demandent ce qu’un résultat de test sur ce gène signifie réellement.
La colle mécanique du cœur
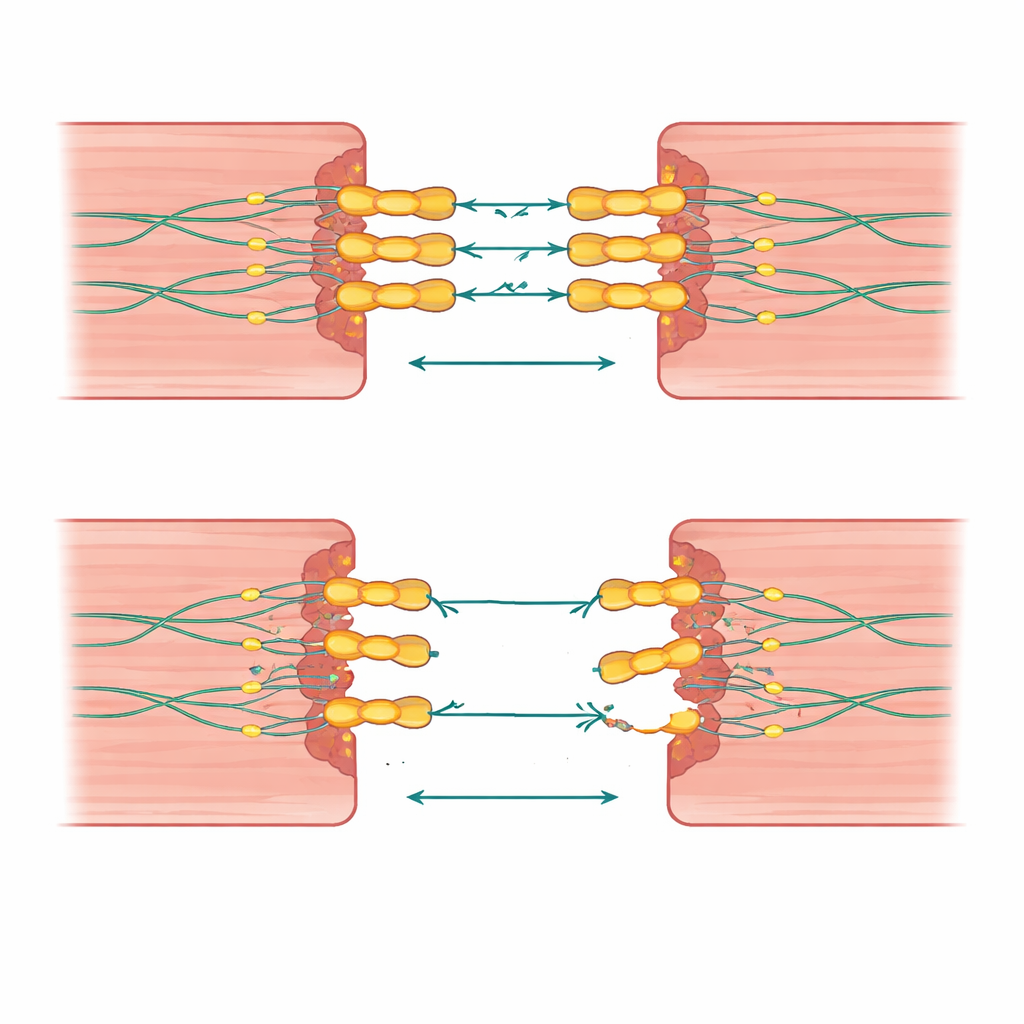
Les cellules du muscle cardiaque doivent rester fermement attachées les unes aux autres tout en battant des millions de fois au cours d’une vie. La desmogleine‑2 fait partie d’une structure microscopique en forme de rivet qui verrouille les cellules voisines afin qu’elles puissent tirer en équipe. Les auteurs expliquent comment cette protéine s’étend de l’extérieur de la cellule, où elle s’accroche à un partenaire correspondant sur la cellule voisine, jusqu’à l’intérieur, où elle s’amarre à un échafaudage de soutien. Étant donné que la desmogleine‑2 est le seul membre de sa famille présent dans les cellules cardiaques, toute atteinte sérieuse ne peut être compensée par un substitut, rendant le cœur particulièrement vulnérable.
Faire la part des variantes significatives et du bruit de fond
Le séquençage moderne identifie des milliers de différences dans le gène de la desmogleine‑2 au sein de la population, mais la plupart ne provoquent pas de maladie. L’équipe a passé en revue de façon systématique 115 études publiées et s’est appuyée sur deux larges bases de données publiques qui répertoriaient ensemble plus de 5 000 variantes. En utilisant des règles de génétique médicale largement acceptées, ils ont reclassé chaque modification selon sa probabilité d’être pathogène. Ils ont constaté que les variants véritablement délétères se regroupent dans des régions spécifiques de la protéine : en particulier les segments externes qui nécessitent du calcium pour former un pont rigide entre les cellules, une courte séquence qui doit être clivée pour que la protéine mûrisse, et la région interne qui s’attache à une autre protéine cardiaque clé. De nombreuses autres modifications sont restées « incertaines », mais un sous‑ensemble a montré de fortes indications d’importance et a été signalé pour un suivi plus rapproché.
Ce que révèle la cohorte italienne
Pour voir comment ces schémas génétiques se traduisent chez de vraies personnes, les chercheurs ont étudié 95 individus en Italie porteurs de variantes de la desmogleine‑2 et évalués en profondeur par imagerie, tests du rythme cardiaque et suivi à long terme. Environ la moitié remplissaient des critères stricts de cardiomyopathie arythmogène, une affection dans laquelle des zones du muscle cardiaque sont progressivement remplacées par de la cicatrice et de la graisse, préparant le terrain pour des troubles du rythme dangereux. Parmi les apparentés porteurs d’une variante, seulement environ quatre sur dix présentaient effectivement des signes de maladie, soulignant qu’un test génétique positif n’entraîne pas automatiquement la maladie mais indique la nécessité d’une surveillance attentive. Les personnes atteintes de la maladie manifeste présentaient une charge notable d’événements rythmiques graves, tandis que les greffes et les décès étaient moins fréquents mais néanmoins présents.
Quand un seul coup ne suffit pas
Un enseignement marquant de ce travail est que le nombre et la combinaison de variantes importent. Les personnes ayant hérité de deux copies défectueuses de la desmogleine‑2, ou d’une variante de desmogleine‑2 plus une modification d’un gène de « colle » cardiaque apparenté, avaient tendance à tomber malades plus jeune et à présenter des atteintes plus étendues des deux côtés du cœur. Certaines familles portaient de grandes délétions ou duplications qui supprimaient ou doublonnaient non seulement la desmogleine‑2 mais aussi des gènes voisins, reliant ces altérations à une maladie agressive et à des foyers de morts subites. En revanche, de nombreux apparentés avec une seule altération affichaient des symptômes légers ou inexistants, ce qui suggère que le contexte génétique et des facteurs de vie tels que l’exercice peuvent faire basculer l’équilibre entre risque silencieux et maladie manifeste.
De la forme de la protéine au risque pour le patient
Pour relier le code ADN aux effets physiques, l’équipe a utilisé des modèles protéiques 3D avancés pour voir comment des substitutions spécifiques pouvaient assouplir l’armature de la desmogleine‑2. Les modifications qui déformaient les boucles de liaison au calcium ou rompaient des points d’attache clés étaient prédites pour déstabiliser la protéine et affaiblir l’adhérence cellule‑à‑cellule. Ces indices structurels ont été réintégrés dans le système de classification, aidant à orienter certaines variantes limites vers une probabilité accrue d’être délétères ou, au contraire, plus probablement bénignes. Ce pont entre modélisation moléculaire et données cliniques fait évoluer le test génétique au‑delà d’une simple lecture du code vers une compréhension plus fonctionnelle.
Ce que cela signifie pour les patients et les familles
Pour les familles touchées par la cardiomyopathie arythmogène, cette étude offre à la fois prudence et orientation. Elle montre que toute variante de la desmogleine‑2 n’est pas une condamnation à une maladie cardiaque sévère, mais que certains schémas — en particulier les atteintes multiples ou les modifications dans des régions critiques de la protéine — sont associés à des problèmes plus précoces et plus graves. Les auteurs soutiennent que les porteurs de ces variantes ne doivent pas être considérés comme « sains tant que l’on n’a pas prouvé le contraire », mais suivis à vie avec des contrôles du rythme et des imageries adaptés. Leur approche intégrative — mêlant génétique à grande échelle, études familiales détaillées et structure protéique — ouvre la voie à des estimations de risque plus précises et à un conseil plus assuré lorsqu’une variante de la desmogleine‑2 apparaît dans un test génétique.
Citation: Pinci, S., Celeghin, R., Martini, M. et al. Integrative genomic and literature assessment of desmoglein 2-related arrhythmogenic cardiomyopathy with Italian cohort validation. Commun Med 6, 145 (2026). https://doi.org/10.1038/s43856-026-01416-w
Mots-clés: cardiomyopathie arythmogène, desmogleine‑2, maladie cardiaque héréditaire, risque génétique, mort subite cardiaque