Clear Sky Science · fr
Diffusion conditionnelle avec alignement modal sensible à la localité pour générer des ensembles conformationnels protéiques diversifiés
Pourquoi le mouvement des protéines compte
Les protéines dans nos cellules ne sont pas des sculptures rigides ; elles se comportent plutôt comme de petites machines flexibles qui changent constamment de forme. Ces changements de conformation peuvent contrôler la façon dont les enzymes catalysent des réactions, la manière dont les récepteurs répondent aux médicaments et la façon dont les signaux circulent dans les cellules. Pourtant, la plupart des images familières de protéines montrent une seule « photo » structurelle, passant à côté de la riche diversité de formes réellement présentes. Cet article présente Mac-Diff, une méthode d’intelligence artificielle capable de générer rapidement de nombreuses formes réalistes pour une protéine donnée, aidant les scientifiques à voir non seulement à quoi ressemble une protéine, mais aussi comment elle respire et bouge.
Des instantanés uniques aux ensembles en mouvement
Pendant des décennies, les chercheurs ont dû s’appuyer sur des expériences laborieuses ou de longues simulations de dynamique moléculaire pour explorer le mouvement des protéines, opérations qui peuvent être lentes et coûteuses. Des outils de rupture comme AlphaFold2 prédisent désormais la structure 3D la plus probable d’une protéine à partir de sa séquence d’acides aminés, mais fournissent généralement une ou quelques conformations privilégiées. De nombreuses protéines, en particulier celles impliquées dans la signalisation et la régulation allostérique, occupent naturellement plusieurs états plus ou moins définis. Les auteurs soutiennent que, pour comprendre comment ces protéines fonctionnent réellement — et pour concevoir des médicaments qui se lient à des formes transitoires moins évidentes — il faut un moyen de générer des ensembles entiers de conformations plausibles, et pas seulement une meilleure estimation unique.
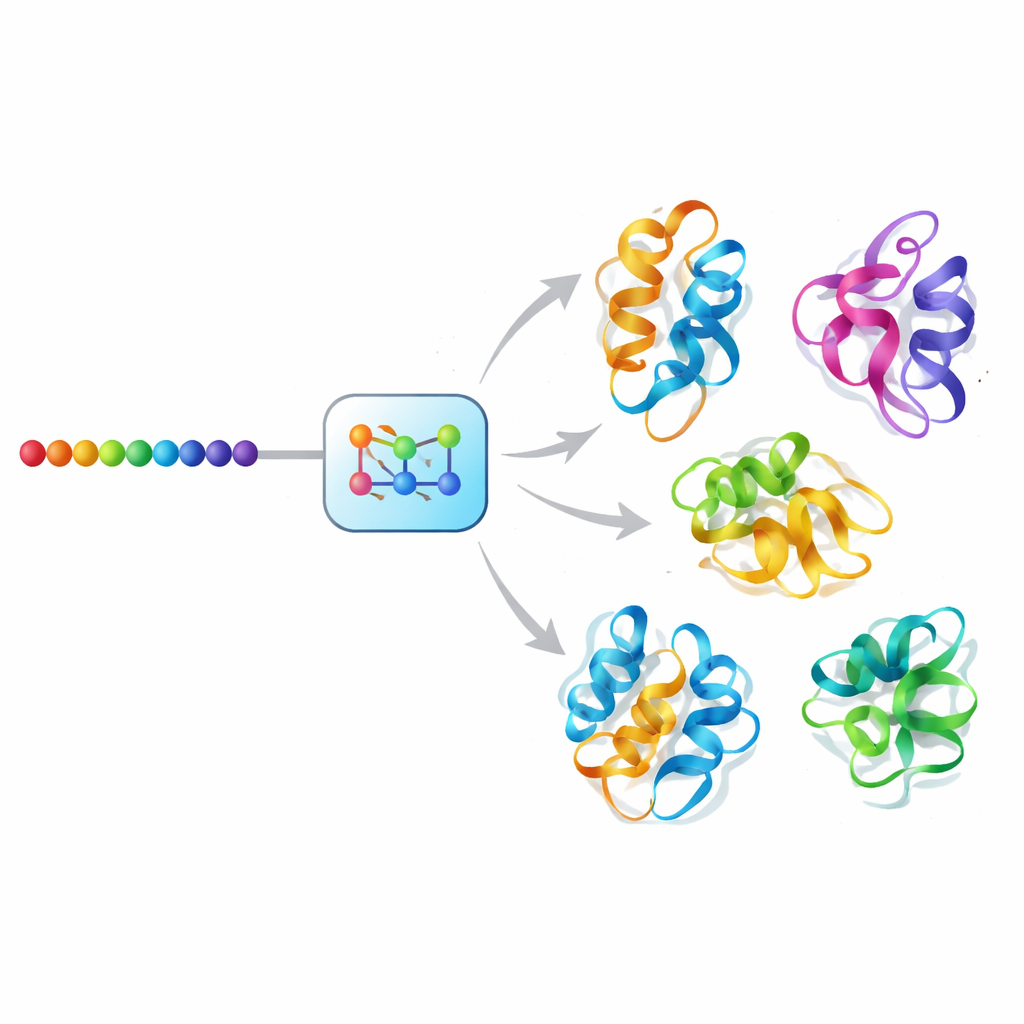
Une approche de « diffusion » par IA pour le mouvement des protéines
Mac-Diff relève ce défi en utilisant un modèle génératif de type diffusion, une classe d’IA qui a permis des avancées récentes en synthèse d’images. Plutôt que de débruiter des photographies, Mac-Diff débruite des descriptions géométriques abstraites des charpentes protéiques. Le modèle représente une protéine comme une matrice de relations par paires entre ses résidus — distances et angles insensibles à la rotation ou à la translation globale de la molécule. Dans une étape directe, le système ajoute progressivement du bruit à ces motifs géométriques jusqu’à ce qu’ils ressemblent à du bruit aléatoire. Dans l’étape inverse, il apprend à retirer le bruit pas à pas, guidé par la séquence d’acides aminés de la protéine, jusqu’à ce que réapparaissent des géométries cohérentes compatibles avec la 3D, qui peuvent ensuite être converties en modèles atomiques complets par des logiciels de construction de structures standard.
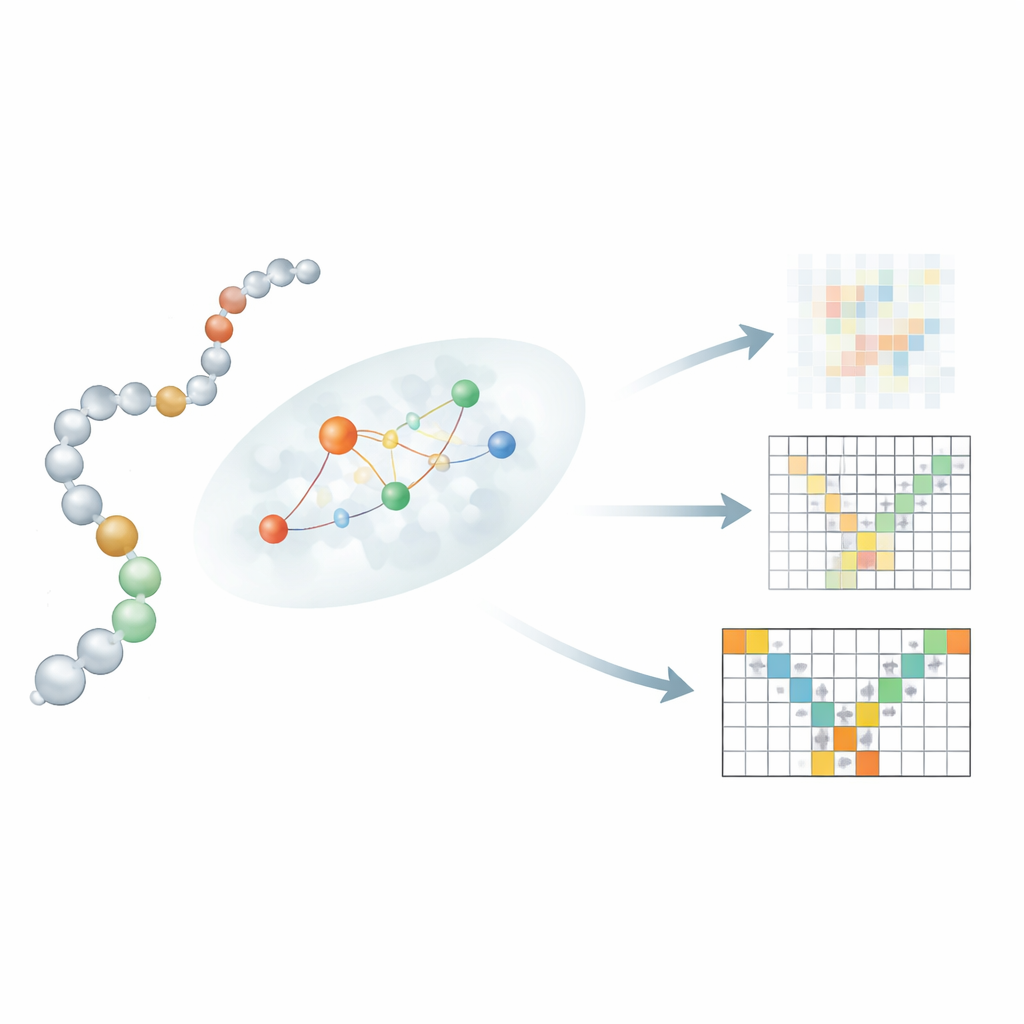
Laisser la séquence dialoguer localement avec la structure
Une innovation clé réside dans la façon dont Mac-Diff relie une séquence linéaire de résidus à leurs voisins 3D. Laisser chaque résidu prêter attention à tous les autres résidus, comme dans les modèles texte‑vers‑image, brouillerait des contraintes physiques importantes. À la place, les auteurs introduisent un mécanisme d’attention « sensible à la localité » qui concentre chaque résidu sur un petit voisinage probable de partenaires d’interaction. Pour estimer ces voisinages, Mac-Diff utilise trois ingrédients : un modèle de langage protéique appelé ESM-2 qui encode le contexte biochimique de chaque résidu ; une carte de contacts suggérant quelles paires de résidus sont susceptibles d’être proches ; et une règle simple favorisant les résidus proches le long de la chaîne. Ces signaux sont combinés de sorte que, pendant le débruitage, le modèle utilise prioritairement l’information provenant des résidus qui sont des partenaires physiquement plausibles, affinant ainsi sa capacité à reconstruire des structures flexibles et réalistes.
Tests face à de longues simulations et à des protéines changeant de forme
Les chercheurs ont évalué Mac-Diff sur deux fronts exigeants. D’abord, ils ont vérifié s’il pouvait reproduire la large distribution de formes observée dans de longues simulations de dynamique moléculaire de haute qualité de protéines à repliement rapide et d’un protéine de référence classique connue sous le nom de BPTI. Sur plusieurs mesures comparant les propriétés statistiques des ensembles générés aux données de simulation — telles que les distributions de distances à l’intérieur de la protéine et la compacité globale — Mac-Diff a égalé ou surpassé les méthodes d’IA concurrentes, tout en générant une plus grande variété de conformations. Il a capturé la plupart des états « métastables » identifiés dans les simulations et a reproduit les schémas de flexibilité au niveau des résidus avec une forte corrélation, indiquant que ses ensembles reflètent de manière réaliste à la fois les plis globaux et les fluctuations locales.
Révéler des états fonctionnels cachés
Ensuite, l’équipe a mis Mac-Diff au défi avec des protéines connues pour adopter des formes très différentes dans l’exercice de leurs fonctions, notamment l’enzyme adénylate kinase, qui bascule entre des formes ouvertes et fermées durant le métabolisme énergétique, ainsi qu’un ensemble soigné de 40 protéines disposant chacune de deux conformations déterminées expérimentalement. Mac-Diff a généré seulement 100 structures candidates par protéine — bien moins que les trajectoires de simulation typiques — et a néanmoins retrouvé la plupart des états connus avec un bon accord géométrique. Pour l’adénylate kinase, par exemple, il a produit à la fois des conformations ouvertes et fermées très similaires aux structures cristallographiques, alors que plusieurs méthodes populaires avaient tendance à favoriser un seul état. Le modèle a également fonctionné environ mille fois plus vite que des simulations conventionnelles sur du matériel comparable, rendant l’exploration systématique de la diversité des formes beaucoup plus pratique.
Qu’est-ce que cela signifie pour la biologie et la médecine
Concrètement, Mac-Diff transforme la séquence d’une protéine en une galerie de poses plausibles plutôt qu’en un portrait unique, et le fait en tenant compte des parties susceptibles de se rapprocher ou de s’agripper en 3D. En échantillonnant ces ensembles avec précision et efficacité, la méthode offre un moyen d’explorer comment de subtiles variations de conformation sous-tendent la fonction, de repérer des conformations rares mais importantes, et de rechercher des poches de liaison pour médicaments qui n’apparaissent que dans des états transitoires. Bien qu’il ne capture pas encore les films ordonnés dans le temps fournis par les simulations, Mac-Diff rend le paysage dynamique des protéines accessible pour beaucoup plus de systèmes, promettant de nouveaux éclairages pour la biologie structurale, la conception de médicaments et l’ingénierie des protéines.
Citation: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
Mots-clés: dynamique des protéines, modèles de diffusion, ensembles conformationnels, protéines allostériques, découverte de médicaments