Clear Sky Science · fr
Robustesse sans précédent des modèles d'énergie atomique informés par la physique à et au‑delà de la température ambiante
Pourquoi cela importe pour la chimie de tous les jours
Les simulations informatiques sont les chevaux de trait de la chimie et des sciences des matériaux modernes. Elles permettent aux scientifiques d’observer des molécules se tordre, vibrer et entrer en collision in silico plutôt que dans des expériences coûteuses et longues. Mais lorsque ces simulations reposent sur de l’apprentissage automatique, elles peuvent soudainement « exploser », produisant des géométries moléculaires impossibles — surtout à des températures élevées. Cette étude présente un nouveau type de modèle d’apprentissage automatique informé par la physique capable d’exécuter de telles simulations pendant des durées extrêmement longues, à des températures allant jusqu’à 1000 kelvins, sans se désintégrer.
Des raccourcis astucieux aux simulations fragiles
La chimie quantique traditionnelle calcule les énergies moléculaires avec une grande précision mais est terriblement lente. Des champs de force plus simples sont rapides mais souvent approximatifs. Les potentiels appris par machine visent à combiner le meilleur des deux mondes : ils apprennent un raccourci reliant la géométrie moléculaire à l’énergie et aux forces, puis utilisent ce raccourci pour piloter la dynamique moléculaire. Sur le papier, beaucoup de ces modèles paraissent excellents, affichant des erreurs moyennes minimes sur des jeux de test standards. En pratique, ces chiffres peuvent être trompeurs. Quand les molécules explorent de nouvelles géométries pendant une simulation — en particulier à haute température — nombre de modèles se retrouvent en dehors de la gamme de structures sur lesquelles ils ont été entraînés. Plutôt que de ramener doucement les molécules vers des formes réalistes, ils peuvent prédire des forces qui étirent ou compressent les liaisons jusqu’à rendre l’ensemble du système non physique et faire planter la simulation.
Construire des modèles à partir d’éléments quantiques
Les auteurs affrontent cette fragilité en modifiant ce que le modèle apprend et la façon dont il est guidé par des connaissances physiques a priori. Ils utilisent un cadre nommé FFLUX, qui s’appuie sur l’approche Interacting Quantum Atoms (IQA). Dans IQA, une molécule est divisée en « atomes topologiques » dont les énergies individuelles sont déterminées directement à partir de la mécanique quantique. Ces énergies atomiques ont une signification physique et s’additionnent pour donner l’énergie totale de la molécule. Plutôt que d’apprendre des énergies de sites arbitraires, les nouveaux modèles à processus gaussiens apprennent ces énergies atomiques d’origine quantique, fournissant une ancre physique profonde pour chaque prédiction. Quatre molécules organiques flexibles — glycine et sérine avec extrémités peptidiques, malondialdéhyde et aspirine — servent de bancs d’essai exigeants en raison de leurs nombreux mouvements internes et de la difficulté connue qu’elles posent aux champs de force appris par machine existants.
Apprendre au modèle à anticiper les problèmes
Une innovation clé réside dans la façon dont le processus gaussien est configuré avant même de voir des données : sa « fonction moyenne », qui encode ce que le modèle suppose dans les régions mal connues. La plupart des travaux antérieurs fixent simplement cette moyenne à zéro, faisant comme si le modèle n’avait aucune attente a priori. Les auteurs décalent délibérément cette moyenne vers des états atomiques d’énergie plus élevée, tout en gardant un comportement physiquement sensé. Ce choix de conception signifie que lorsque le modèle doit extrapoler — par exemple quand des liaisons sont temporairement trop étirées — il favorise naturellement des prédictions qui pénalisent les distorsions extrêmes. Dans des tests étendus, des versions du modèle ne différant que par ce choix de prior ont eu des comportements très différents. Les modèles avec des moyennes conventionnelles ou de basse énergie survivent souvent moins d’un picoseconde avant qu’une molécule n’explose ou ne s’effondre. En revanche, la meilleure moyenne haute énergie (appelée MF5) a permis des simulations stables pendant la fenêtre complète d’un nanoseconde aux températures de 300 à 1000 kelvins pour les quatre molécules.
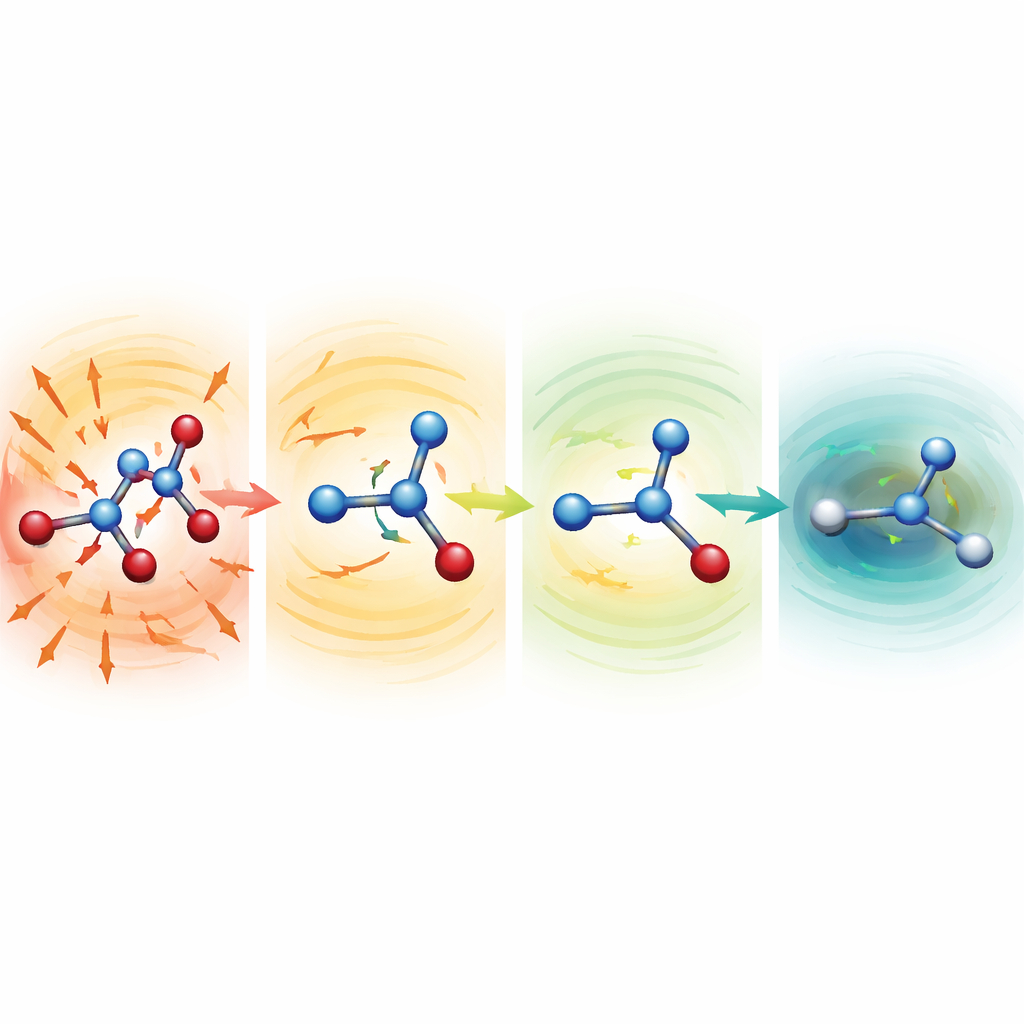
Voir les molécules déformées se réparer
Pour comprendre pourquoi les modèles robustes fonctionnent si bien, les chercheurs ont démarré des simulations à partir de structures volontairement malmenées, avec des liaisons fortement étirées ou comprimées. Pour la sérine, l’aspirine et le malondialdéhyde, ces points de départ se situaient à plusieurs centaines voire plus de mille kilocalories par mole au‑dessus d’une structure normale — des configurations normalement catastrophiques. Avec des fonctions moyennes plus faibles, les molécules se disloquaient rapidement. Avec la configuration MF5, cependant, les forces prédites indiquaient immédiatement des directions qui raccourcissaient les liaisons allongées et allongeaient celles écrasées. En quelques dizaines à quelques centaines de pas de temps, les molécules se relaxaient vers des formes réalistes puis continuaient d’évoluer de façon stable. L’équipe a aussi montré que, sans jamais être entraînés sur des forces, les mêmes modèles peuvent guider des optimisations de géométrie de la dipeptide alanine, reproduisant des conformations à basse énergie connues et des énergies relatives à quelques dixièmes de kilocalorie par mole près, mais à un coût environ 200 fois plus faible que les calculs quantiques complets.
Simulations longues et chaudes sur du matériel courant
La robustesse ne consiste pas seulement à survivre à un départ difficile ; il s’agit de tenir sur des millions ou des milliards de pas de temps. Les auteurs ont poussé leurs meilleurs modèles plus loin en lançant 50 simulations indépendantes à 500 kelvins, chacune durant 10 nanosecondes, pour leurs quatre molécules test. Aucune de ces exécutions n’a planté, totalisant un temps de simulation combiné d’un demi‑microseconde — inhabituel pour des champs de force appris par apprentissage automatique à la pointe. Fait encore plus marquant, les simulations ont tourné efficacement sur des CPU standards, rivalisant pas à pas avec, voire surpassant, certains potentiels à réseau de neurones exigeant des GPU puissants. Tout au long des essais, les molécules ont exploré des ensembles riches de formes et d’états métastables, montrant que la robustesse n’a pas été obtenue en bloquant artificiellement les mouvements ou en imposant des structures rigides.
Ce que cela signifie pour la modélisation moléculaire future
Pour les non‑spécialistes, le message principal est que tous les modèles d’apprentissage automatique à faible erreur ne sont pas fiables lorsque l’on pousse les systèmes hors de leurs habitudes. En ancrant leurs modèles dans des énergies atomiques dérivées de la mécanique quantique et en ajustant soigneusement les attentes intégrées du modèle vers des états de haute énergie, les auteurs ont créé une famille de potentiels qui produisent naturellement des « forces de rappel » — l’équivalent moléculaire d’un harnais de sécurité — qui maintiennent les simulations physiques même à haute température et à partir de points de départ déformés. Cette approche promet des simulations de molécules complexes plus longues et plus fiables, et ouvre la voie à des extensions futures où des modèles similaires, informés par la physique, traiteront des phases condensées et d’interactions subtiles comme la dispersion, tout en restant économiquement viables en termes de calcul.
Citation: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Mots-clés: champs de force par apprentissage automatique, robustesse en dynamique moléculaire, potentiels à processus gaussiens, modélisation informée par la physique, énergies atomiques quantiques