Clear Sky Science · fr
Affinage d’AlphaFold avec des observations cryo-EM limitées
Pourquoi il est si difficile de voir la forme des protéines
Les protéines sont de minuscules machines moléculaires qui pilotent presque tous les processus de notre organisme, de la production d’énergie à la transmission des signaux nerveux. Pour comprendre leur fonctionnement — et comment des médicaments pourraient les moduler — les scientifiques doivent connaître leurs formes tridimensionnelles précises. Deux outils puissants se sont imposés pour cette tâche : la cryo‑microscopie électronique (cryo‑EM), qui prend de nombreux clichés flous de protéines congelées, et AlphaFold, un système d’intelligence artificielle qui prédit les structures protéiques à partir de leurs séquences. Mais dans beaucoup d’expériences réelles, les données cryo‑EM sont incomplètes, et les prédictions d’AlphaFold ne correspondent pas toujours à la réalité. Cet article présente CoCoFold, une méthode qui apprend à AlphaFold à tenir compte directement des données cryo‑EM difficiles et à améliorer ses prédictions en conséquence.
Quand l’appareil photographie trop peu
La cryo‑EM consiste à congeler instantanément les protéines et à imager d’énormes nombres de particules individuelles sous de nombreux angles, puis à combiner ces images en une carte 3D. En pratique, cependant, les chercheurs ne disposent souvent pas d’assez d’images exploitables. Parfois la protéine n’existe que brièvement dans un état à haute énergie, si bien que très peu de particules sont capturées. Dans d’autres cas, les protéines privilégient certaines orientations à la surface de la vitre, si bien que de nombreux angles de vue sont manquants. Ces deux problèmes conduisent à des cartes floues et incomplètes difficiles à traduire en modèles atomiques fiables. Les logiciels existants peuvent ajuster les structures prédites par AlphaFold dans de telles cartes, mais leur succès dépend fortement de la disponibilité initiale de données nettes et à haute résolution.
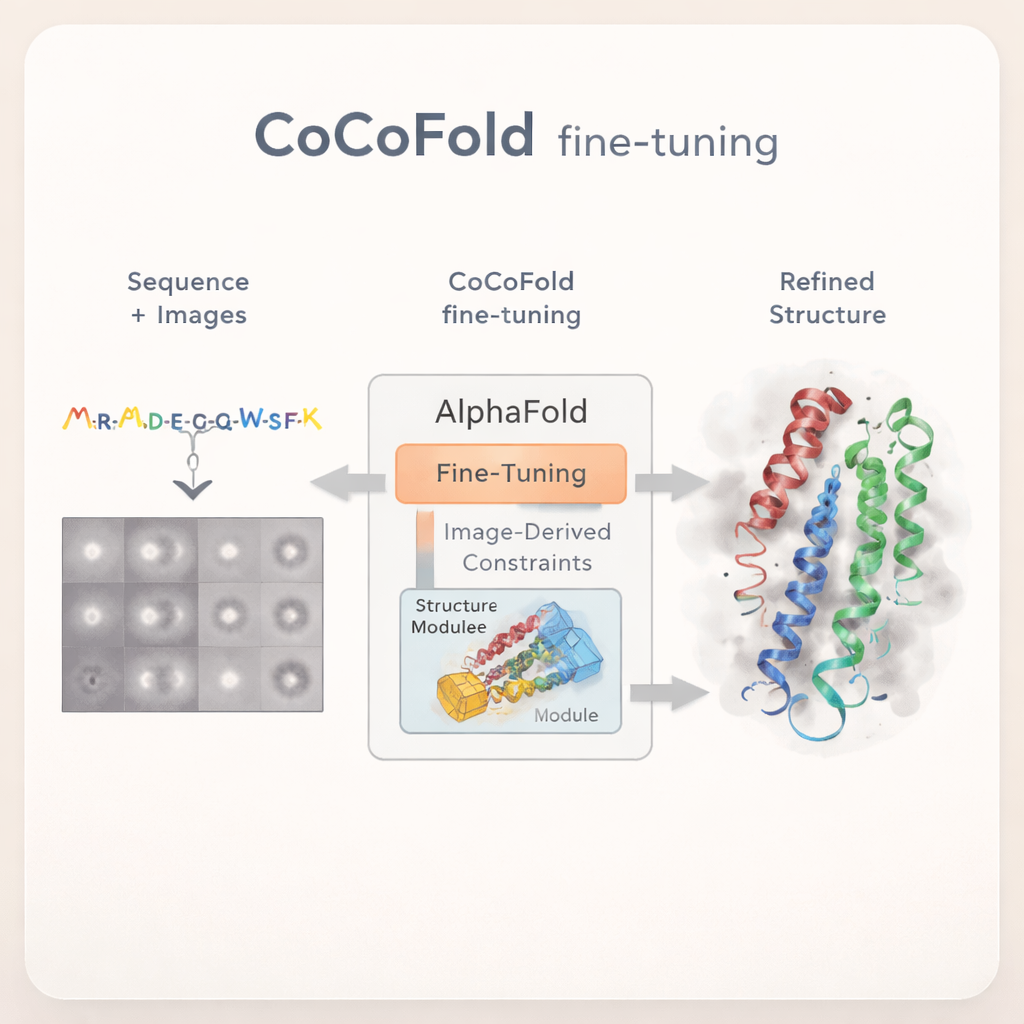
Apprendre à AlphaFold à partir des images brutes
CoCoFold adopte une approche différente : au lieu de s’appuyer sur une carte 3D cryo‑EM entièrement reconstruite, il utilise directement les images 2D brutes des particules pour affiner AlphaFold. La méthode part d’une prédiction AlphaFold‑Multimer et laisse gelée la majeure partie du réseau original, préservant ainsi sa vaste connaissance du repliement des protéines. Seule la dernière partie de construction de la structure est autorisée à évoluer. Un « adaptateur » léger est ajouté pour injecter dans ce module structural l’information dérivée des images cryo‑EM, orientant doucement le modèle vers des formes compatibles avec les données expérimentales tout en évitant des déviations incompatibles avec la physique des protéines connue.
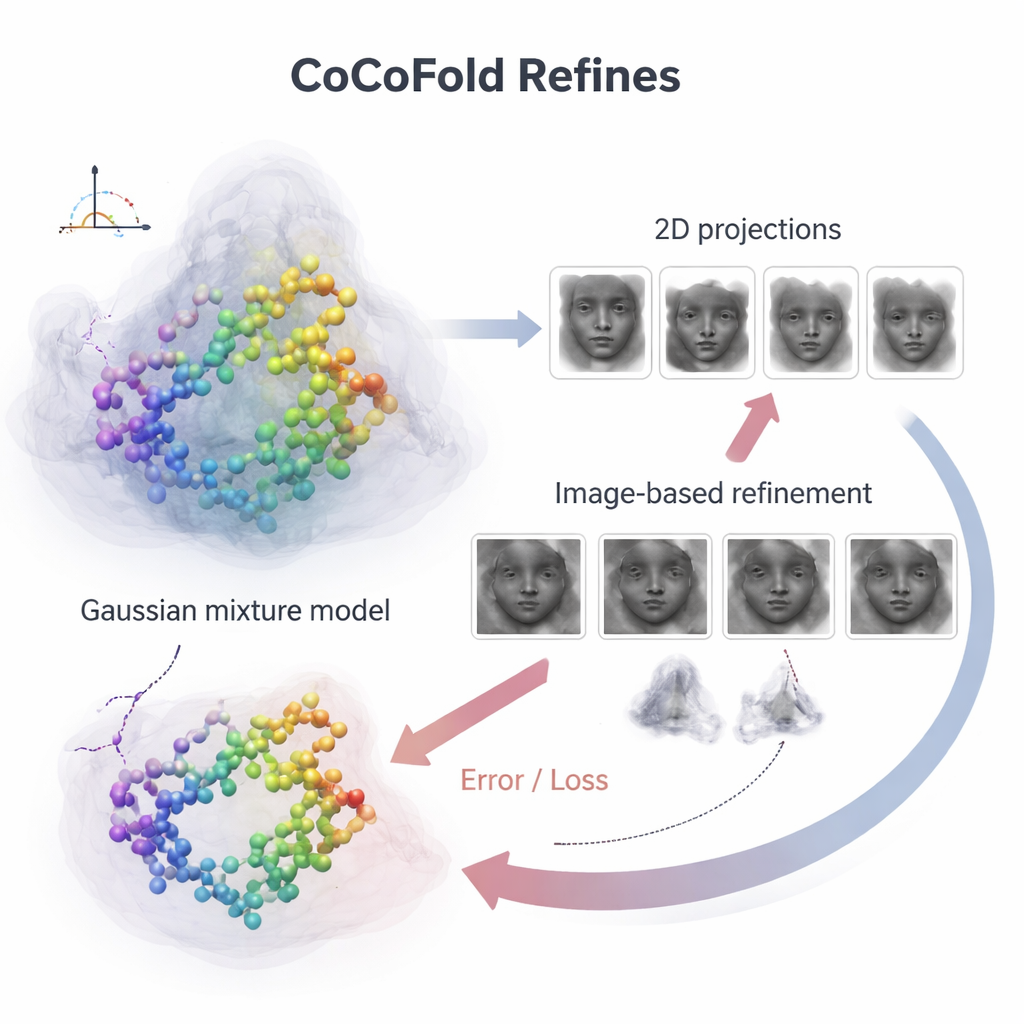
Transformer des images en rétroaction structurale
Pour relier les atomes individuels de la protéine aux images bruyantes du microscope, CoCoFold construit une représentation lisse et flexible de la structure prédite à l’aide de blobs tridimensionnels qui se chevauchent, appelée mélange gaussien. À partir de cette représentation, il simule l’apparence de la protéine dans le microscope aux mêmes directions de vue et conditions d’imagerie que dans l’expérience réelle. Ces clichés simulés sont ensuite comparés aux particules cryo‑EM réelles, anneau par anneau dans le domaine des fréquences, pour évaluer leur concordance. Tout désaccord devient un signal de rétroaction qui est renvoyé à travers le réseau, ajustant légèrement à la fois le modèle protéique et la représentation de densité. Après l’entraînement, le modèle atomique est en outre épuré par une étape de raffinage basée sur la physique afin d’éliminer les conflits géométriques locaux.
Rester précis quand les données sont rares ou biaisées
Les auteurs ont testé CoCoFold sur plusieurs jeux de données expérimentaux et simulés conçus pour reproduire les deux principaux problèmes de la cryo‑EM : trop peu de particules et de larges lacunes dans les angles de vue. Dans ces conditions difficiles, les outils standards — y compris d’autres méthodes d’apprentissage profond qui dépendent de cartes reconstruites — avaient tendance à perdre des régions de la protéine, à mal positionner des hélices ou à perdre des détails fins à mesure que les cartes s’estompaient. CoCoFold, en revanche, produisait systématiquement des modèles correspondant de manière plus fidèle et plus complète aux structures de référence. Ses erreurs restaient faibles même lorsque le nombre de particules était drastiquement réduit ou lorsque de larges cônes de directions de vue manquaient, ce qui suggère que l’apprentissage direct à partir des images brutes préserve des informations cruciales que les approches basées sur les cartes jettent.
Ce que cela signifie pour la biologie structurale à venir
Pour les non‑spécialistes, le message clé est que CoCoFold agit comme un traducteur entre les prédictions puissantes de l’IA et les données expérimentales imparfaites. Plutôt que de faire aveuglément confiance à AlphaFold ou à la cryo‑EM, il permet aux deux de s’informer mutuellement, en particulier dans les régimes difficiles où les expériences ne fournissent qu’une vue partielle. Dans les cas simples avec des données abondantes et de haute qualité, les outils classiques basés sur les cartes restent très performants. Mais lorsque les particules sont rares ou que des orientations font défaut — situations courantes lors de l’étude d’états protéiques fugitifs ou fragiles — CoCoFold offre un moyen de retrouver des modèles atomiques fiables à partir d’informations qui, autrement, seraient perdues.
Citation: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Mots-clés: cryo-EM, AlphaFold, structure des protéines, apprentissage profond, biologie structurale