Clear Sky Science · fr
L’échec énergétique mitochondrial sous-tend la neuropathie sensitive liée à FLVCR1
Quand les nerfs de la douleur manquent d’énergie
Certaines personnes naissent presque incapables de ressentir la douleur. À première vue, cela peut sembler une bénédiction, mais cela devient vite une malédiction : sans douleur comme signal d’alerte, elles accumulent brûlures, fractures, infections et parfois cécité. Cette étude examine une forme rare de ces troubles de perte de douleur et met en lumière un coupable surprenant : de minuscules centrales électriques à l’intérieur des cellules nerveuses dont la production d’énergie se dérègle gravement.
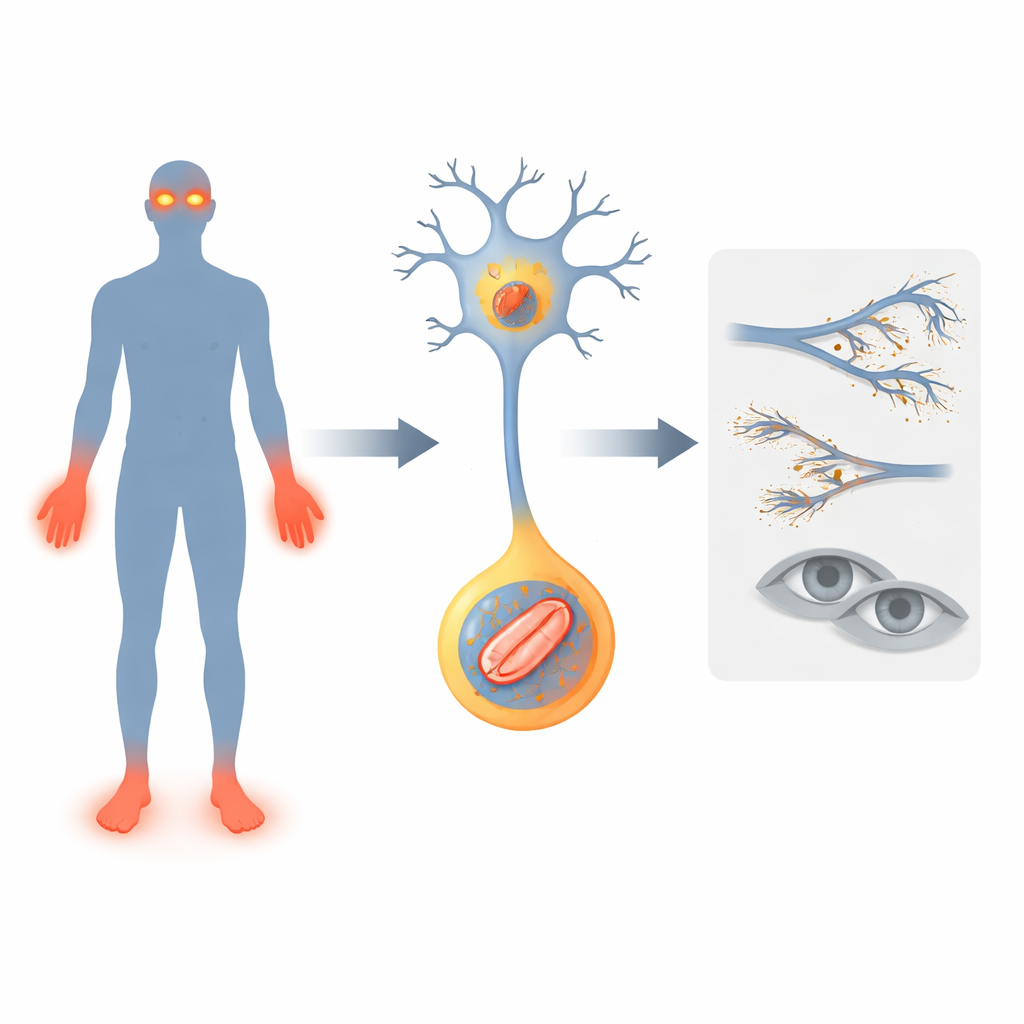
Un gène qui fait taire les sonnettes d’alarme
Les chercheurs se concentrent sur un gène nommé FLVCR1, déjà associé à des affections nerveuses rares où les personnes perdent la sensation de douleur, développent une démarche instable et parfois une perte progressive de la vision. Ils décrivent deux nouveaux patients porteurs de variantes de FLVCR1. Les deux enfants présentaient des signes précoces : retard des acquisitions motrices, chutes fréquentes, infections profondes et mutilations des doigts et des orteils parce que les blessures restaient inaperçues. L’un d’eux a aussi développé une maladie dégénérative de l’œil, la rétinite pigmentaire, entraînant une cécité nocturne. Ces cas élargissent le spectre des manifestations des défauts de FLVCR1 chez l’humain et renforcent l’idée que ce gène est essentiel au maintien en vie des neurones sensoriels de la douleur et des cellules photoréceptrices de la rétine.
Modéliser la maladie chez de petits poissons
Pour explorer comment FLVCR1 affecte le développement des nerfs sensitifs, l’équipe s’est tournée vers le poisson-zèbre, dont les embryons transparents permettent d’observer directement les cellules nerveuses. Ils ont réduit les niveaux de la version du gène chez le poisson, flvcr1a, à l’aide d’outils génétiques. Les poissons avec une expression diminuée de flvcr1a présentaient moins de ganglions rachidiens dorsaux, des amas de neurones qui détectent le toucher et la douleur le long de la colonne. Au plan comportemental, ces poissons bougeaient moins spontanément et ne nageaient que de courtes distances quand leur queue était doucement stimulée, suggérant une réponse sensorielle atténuée. Comme les modèles murins antérieurs mouraient trop tôt pour analyser leurs nerfs sensitifs, ces poissons-zèbres fournissent le premier système vivant dans lequel les défauts nerveux et comportementaux liés à FLVCR1 peuvent être suivis en détail.
Trois voies perturbées convergent vers les centrales cellulaires
FLVCR1 se situe dans les membranes cellulaires et gère plusieurs substances clés. Des travaux antérieurs suggéraient des rôles dans la gestion de la choline (un composant des lipides membranaires), de l’hème (le pigment contenant du fer qui alimente de nombreuses enzymes) et du flux calcique entre compartiments cellulaires. Les scientifiques ont prélevé des cellules de peau (fibroblastes) chez quatre patients portant différentes mutations de FLVCR1 et les ont comparées à des cellules de personnes saines et de porteurs asymptomatiques. Ils ont constaté que les cellules des patients avaient des niveaux de choline plus faibles et des membranes cellulaires plus fluides, des changements susceptibles de perturber l’environnement lipidique délicat requis par les mitochondries, les organites producteurs d’énergie. Ils ont aussi découvert qu’une enzyme cruciale pour la synthèse d’hème dans les mitochondries, ALAS1, était moins active, même si la teneur totale en hème paraissait presque normale. Parallèlement, les sites de contact physiques entre le réticulum endoplasmique et les mitochondries — où le calcium entre normalement dans les mitochondries — étaient plus courts et moins fréquents, et l’entrée de calcium dans les mitochondries était réduite. Trois problèmes — pénurie de choline, synthèse d’hème ralentie et transfert calcique affaibli — pointent tous vers une performance mitochondriale altérée.
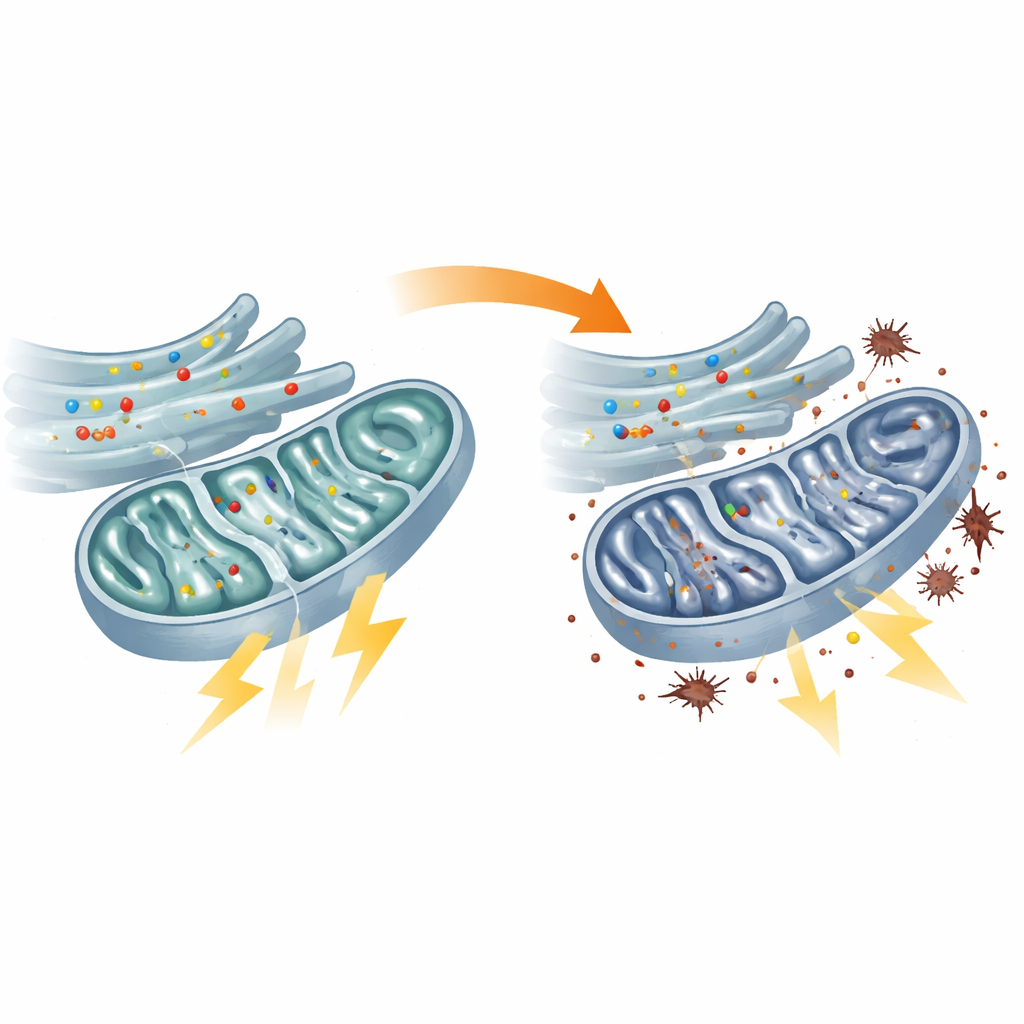
Mitochondries affamées et systèmes de secours surmenés
Des tests directs du métabolisme énergétique ont confirmé que les mitochondries des fibroblastes des patients fonctionnaient en dessous de leur capacité. Le carrefour central de la combustion des carburants, le cycle TCA, était plus lent, plusieurs de ses enzymes clés étaient moins actives et la chaîne de réactions qui convertit normalement le carburant en ATP, la monnaie énergétique de la cellule, était affaiblie. En conséquence, le niveau d’ATP à l’intérieur des mitochondries chutait. Les cellules ont tenté de compenser en augmentant la glycolyse, une voie de dégradation du sucre moins efficace, hors des mitochondries. Ce changement de stratégie énergétique a un effet secondaire : des électrons fuyaient des systèmes mitochondriaux stressés et provoquaient des niveaux accrus de peroxydation lipidique, une forme de dommage oxydatif aux membranes cellulaires. Des défauts similaires ont été observés chez les poissons-zèbres à flvcr1a réduit, reliant directement la défaillance mitochondriale au modèle animal de neuropathie sensitive.
Indices de traitements futurs par renforcement énergétique cellulaire
De manière encourageante, certaines de ces anomalies pouvaient être atténuées en laboratoire. Lorsque l’équipe a augmenté artificiellement l’entrée de calcium dans les mitochondries en surexprimant une protéine canal appelée MCU dans des cellules de patients, la production d’énergie a rebondi et les signes de dommages oxydatifs ont diminué. Fournir aux cellules un précurseur de l’hème, l’acide 5-aminolévulinique (ALA), a également amélioré l’activité du cycle TCA, la fonction de la chaîne respiratoire et les niveaux d’ATP, bien qu’une exposition prolongée à l’ALA ait montré des effets délétères dans des études antérieures. Un apport supplémentaire en choline a normalisé la fluidité membranaire et contribué à réduire les dommages lipidiques, mais n’a procuré que des gains énergétiques modestes et à court terme. Ces expériences de « sauvetage » suggèrent qu’aucune voie unique n’est seule responsable ; c’est plutôt un réseau perturbé de gestion de la choline, de l’hème et du calcium qui pousse les mitochondries vers une sous-performance chronique.
Pourquoi ces résultats comptent pour les patients
En retraçant les conséquences des mutations de FLVCR1 depuis les molécules jusqu’aux cellules et aux organismes entiers, ce travail propose que la défaillance énergétique mitochondriale est une force motrice derrière cette forme de neuropathie de perte de la douleur et ses problèmes visuels associés. Les nerfs sensitifs et les photorécepteurs ont des besoins énergétiques exceptionnellement élevés parce qu’ils maintiennent de longs axones ou renouvellent en continu des structures sensibles à la lumière, les rendant particulièrement vulnérables lorsque la production mitochondriale faiblit. Le modèle poisson-zèbre de l’étude et les cellules dérivées de patients offrent désormais des terrains d’essai pratiques pour des thérapies visant à renforcer le métabolisme mitochondrial. Bien que des traitements tels que la supplémentation en choline, une stimulation contrôlée de la synthèse d’hème ou des médicaments augmentant l’entrée de calcium mitochondrial nécessitent une évaluation soigneuse dans des modèles animaux et des essais cliniques, le message principal est clair : restaurer l’alimentation électrique de neurones fragiles pourrait un jour aider à protéger des personnes nées sans le signal d’alerte le plus important de la nature — la douleur.
Citation: Bertino, F., Zanin Venturini, D.I., Grasso, E. et al. Mitochondrial energetic failure underlies FLVCR1-related sensory neuropathy. Commun Biol 9, 429 (2026). https://doi.org/10.1038/s42003-026-09691-y
Mots-clés: neuropathie sensitive, dysfonction mitochondriale, FLVCR1, insensibilité à la douleur, métabolisme énergétique nerveux