Clear Sky Science · fr
Un guide pratique des technologies de séquençage ciblé de l’ARN en cellule unique
Pourquoi examiner les cellules une par une importe
Toutes les cellules de votre corps possèdent le même ADN, et pourtant elles se comportent très différemment. Elles y parviennent en activant ou en désactivant des gènes spécifiques et en modifiant les molécules d’ARN de façons subtiles. Le séquençage moderne de l’ARN en cellule unique peut lire quels ARNs sont présents dans des milliers de cellules simultanément, mais il manque actuellement la majeure partie du message. Cette revue explique où les techniques actuelles perdent de l’information et comment de nouvelles méthodes « ciblées » sont développées pour zoomer sur les parties les plus importantes des molécules d’ARN pour la recherche, le diagnostic et la conception de traitements.
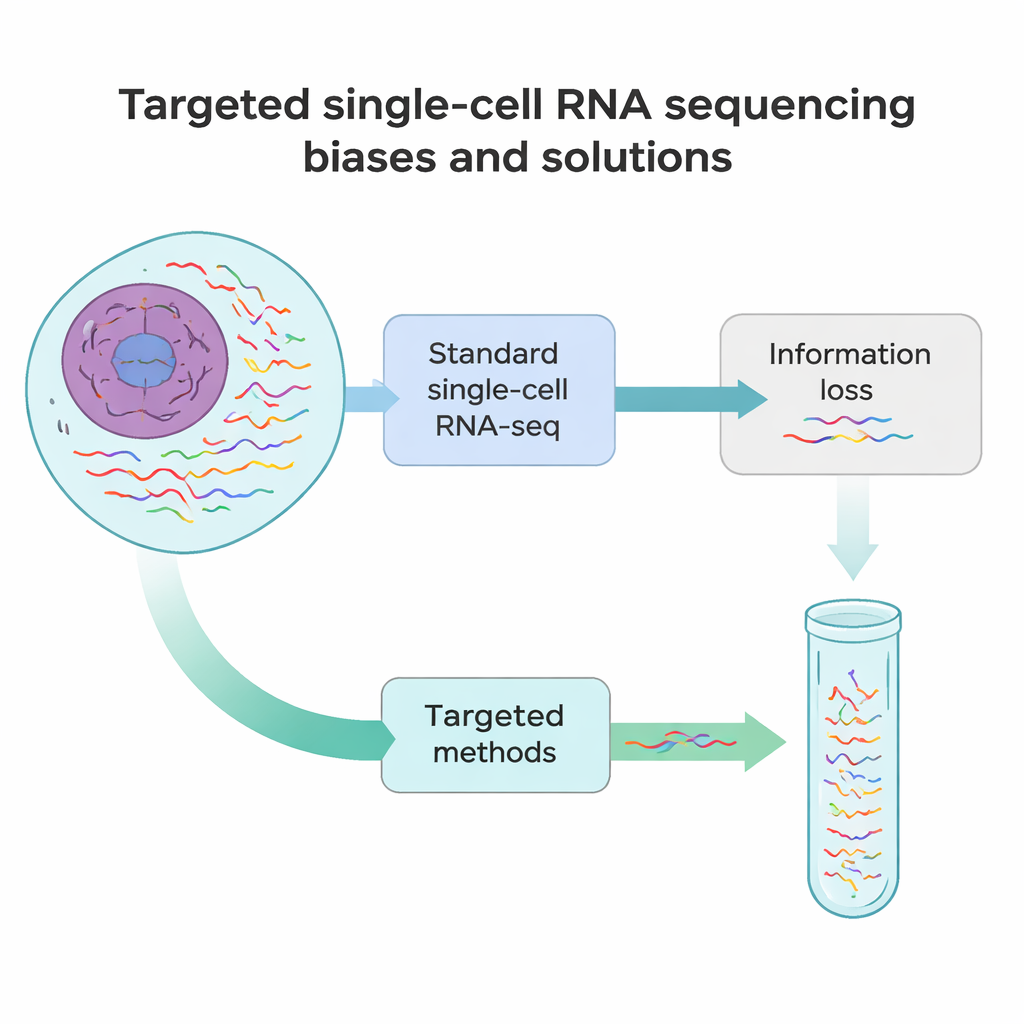
Où les méthodes actuelles montrent leurs limites
Le séquençage standard de l’ARN en cellule unique fonctionne un peu comme prendre un instantané rapide de chaque message plutôt qu’un film en longueur. Dans la plupart des expériences, seules environ 10–40 % de tous les ARNs d’une cellule sont détectés, et on ne lit que leur début ou leur fin. Cela signifie que de nombreux ARNs rares mais importants — comme des marqueurs définissant l’identité d’une cellule, ou des versions de gènes portant des mutations responsables de maladies — sont facilement manqués. De plus, plusieurs étapes techniques, depuis la dissociation des tissus en cellules uniques jusqu’à la copie de l’ARN en ADN et son amplification, introduisent des biais systématiques. Certains ARNs sont tronqués prématurément, d’autres sont surreprésentés, et d’autres encore disparaissent complètement des données.
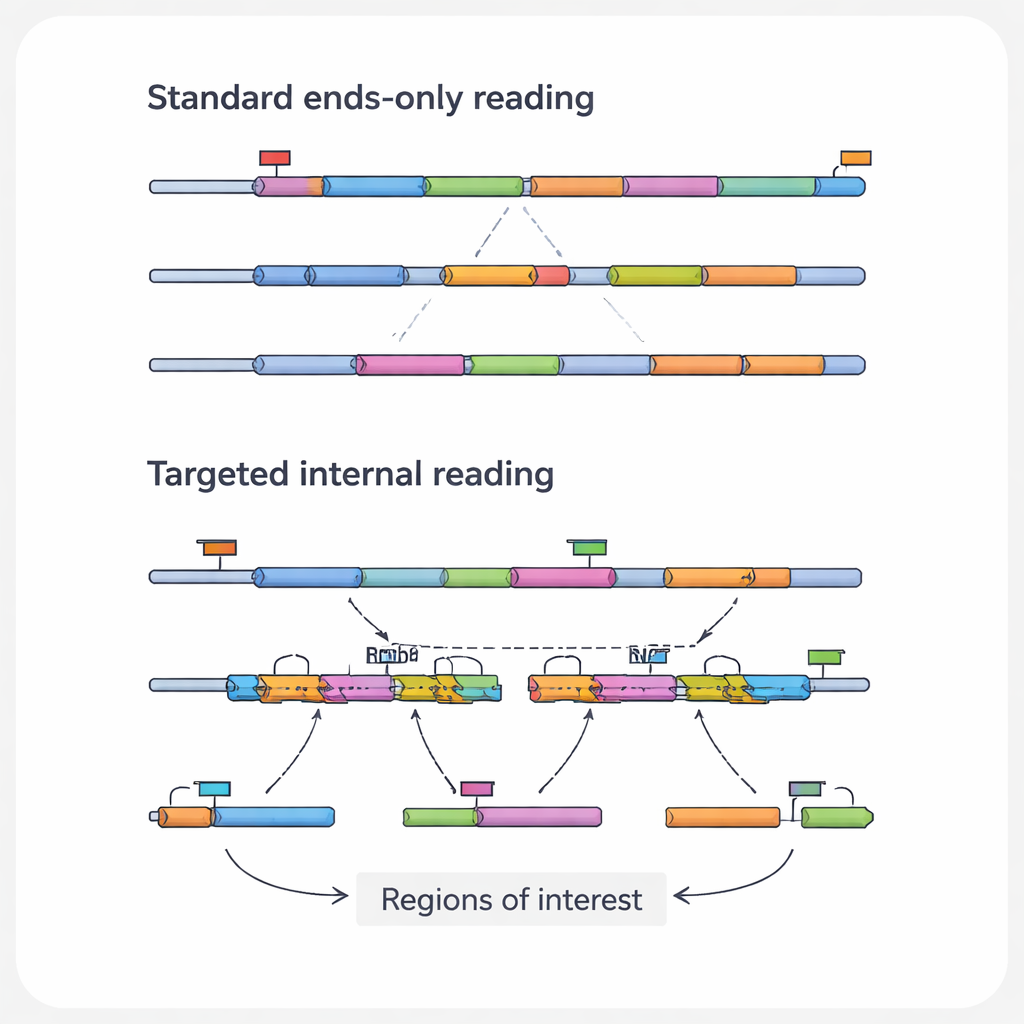
Pourquoi les détails internes de l’ARN comptent
Les informations les plus pertinentes médicalement dans une molécule d’ARN se trouvent souvent dans ses régions internes, et non aux extrémités que voient les méthodes standard. Ces sections internes peuvent contenir des mutations ponctuelles qui entraînent un cancer, des points de fusion où deux gènes se sont anormalement reliés, ou des jonctions d’épissage qui créent différentes variantes protéiques à partir d’un même gène. Elles peuvent aussi garder la trace des interventions d’outils d’édition génétique tels que CRISPR. Les auteurs appellent ces caractéristiques spécifiques « régions d’intérêt », et les ARNs qui les portent « transcrits d’intérêt ». Parce que les plateformes à haut débit courantes lisent principalement les extrémités des ARNs, elles négligent régulièrement ces détails cruciaux, en particulier dans les transcrits longs ou de faible abondance.
Nouvelles façons de viser le projecteur
Pour surmonter ces angles morts, les chercheurs ont développé une famille d’approches de séquençage ciblé de l’ARN en cellule unique. Plutôt que d’essayer de lire chaque ARN de manière égale, ces méthodes enrichissent délibérément certains transcrits ou régions sélectionnés. Certaines stratégies repensent les billes de capture pour qu’elles s’attachent à des séquences internes d’ARN plutôt qu’au simple A‑poly(A) de la queue, attirant les messages choisis dès la première étape de constitution de la bibliothèque. D’autres ajoutent des amorces personnalisées qui commencent la copie à un point interne, ou des étapes PCR supplémentaires qui amplifient spécifiquement une liste restreinte de gènes à partir d’une bibliothèque existante. Un autre groupe utilise des sondes en ADN qui s’hybrident aux ARNs cibles ou à leurs copies puis les récupèrent, souvent à l’aide d’étiquettes chimiques simples. Chaque catégorie fait des compromis entre sensibilité, nombre de cellules, nombre de cibles et coût, mais toutes poursuivent le même objectif : récupérer plus de détails signifiants avec le même nombre ou moins de lectures de séquençage.
Applications, des virus aux tumeurs
Ces méthodes ciblées transforment déjà plusieurs domaines de la biologie et de la médecine. En infections, elles peuvent enfin capturer des ARNs viraux ou bactériens dépourvus des queues poly(A) que les protocoles standards attendent, révélant quelles cellules hôtes ils infectent et comment ils modifient l’activité génique de l’hôte. En cancérologie, le séquençage ciblé en cellule unique peut identifier précisément quels types cellulaires portent des mutations ou des gènes de fusion spécifiques et relier ces altérations à des programmes géniques modifiés, aidant à expliquer pourquoi certaines cellules deviennent résistantes aux traitements. D’autres méthodes se concentrent sur l’épissage alternatif, dévoilant quels types cellulaires utilisent quelles isoformes, ou sur des populations cellulaires rares et des marqueurs subtils qui resteraient autrement indétectables. Dans les criblages CRISPR en pool, une meilleure capture des ARN guides permet de relier chaque perturbation génétique à sa réponse cellulaire précise.
Choisir le bon outil et perspectives
Comme il existe désormais une boîte à outils dense d’approches ciblées, les auteurs proposent un arbre de décision pour aider les chercheurs à choisir une méthode. Les questions clés incluent la nécessité d’un profilage du transcriptome complet, le nombre de gènes ou de régions à cibler, la distance de ces régions par rapport aux extrémités de l’ARN, et le nombre de cellules pouvant être traitées. Pour l’avenir, ils estiment que les gains les plus importants viendront de l’amélioration des toutes premières étapes de capture, de l’expansion des stratégies ingénieuses basées sur des sondes, et de la combinaison du ciblage avec les plateformes émergentes de séquençage longue lecture et de séquençage direct de l’ARN. Tant qu’il ne sera pas pratique de lire chaque ARN de chaque cellule de bout en bout, le séquençage ciblé de l’ARN en cellule unique restera essentiel pour voir les parties du message cellulaire qui comptent le plus pour la biologie et la maladie.
Citation: Moro, G., Brunner, E. & Basler, K. A practical guide to targeted single-cell RNA sequencing technologies. Commun Biol 9, 250 (2026). https://doi.org/10.1038/s42003-026-09675-y
Mots-clés: séquençage de l’ARN en cellule unique, séquençage ciblé, transcriptomique, mutations cancéreuses, transcriptomique spatiale