Clear Sky Science · fr
La déméthylase des histones KDM7A régule négativement la polarisation des macrophages profibrotiques et la progression de la fibrose pulmonaire
Pourquoi les poumons cicatrisés nous concernent tous
Lorsque les poumons développent des cicatrices persistantes, respirer devient une lutte quotidienne. Cette affection, appelée fibrose pulmonaire, touche des millions de personnes et n’a actuellement pas de cure — seulement des médicaments qui ralentissent les dégâts. Dans cette étude, les chercheurs mettent au jour un « frein » moléculaire jusque-là caché dans des cellules immunitaires appelées macrophages, qui aide à limiter la formation de cicatrices pulmonaires. Comprendre ce frein pourrait ouvrir la voie à de nouveaux traitements non seulement pour la fibrose pulmonaire, mais aussi potentiellement pour d’autres maladies où cicatrisation nuisible et inflammation incontrôlée vont de pair.
Le récit de cellules immunitaires métamorphes
Les macrophages sont des cellules immunitaires de première ligne qui patrouillent les tissus, éliminent les débris et participent à la réparation des lésions. Mais ce sont aussi des métamorphes : dans certaines situations, ils deviennent des effecteurs pro-inflammatoires, tandis que dans d’autres ils se transforment en réparateurs qui peuvent favoriser la formation de cicatrices. Un type particulier favorisant la cicatrisation, appelé macrophages profibrotiques (Fib-Mac), est fortement associé à la fibrose pulmonaire. Ces cellules produisent des molécules qui activent les fibroblastes, lesquels déposent alors un excès de collagène et d’autres composants de matrice, raidir progressivement le poumon. Les auteurs ont voulu savoir comment les « réglages » génétiques à l’intérieur des macrophages déterminent s’ils deviennent ces cellules Fib-Mac dangereuses ou restent dans des états plus équilibrés et protecteurs.

Un frein épigénétique dissimulé dans le génome
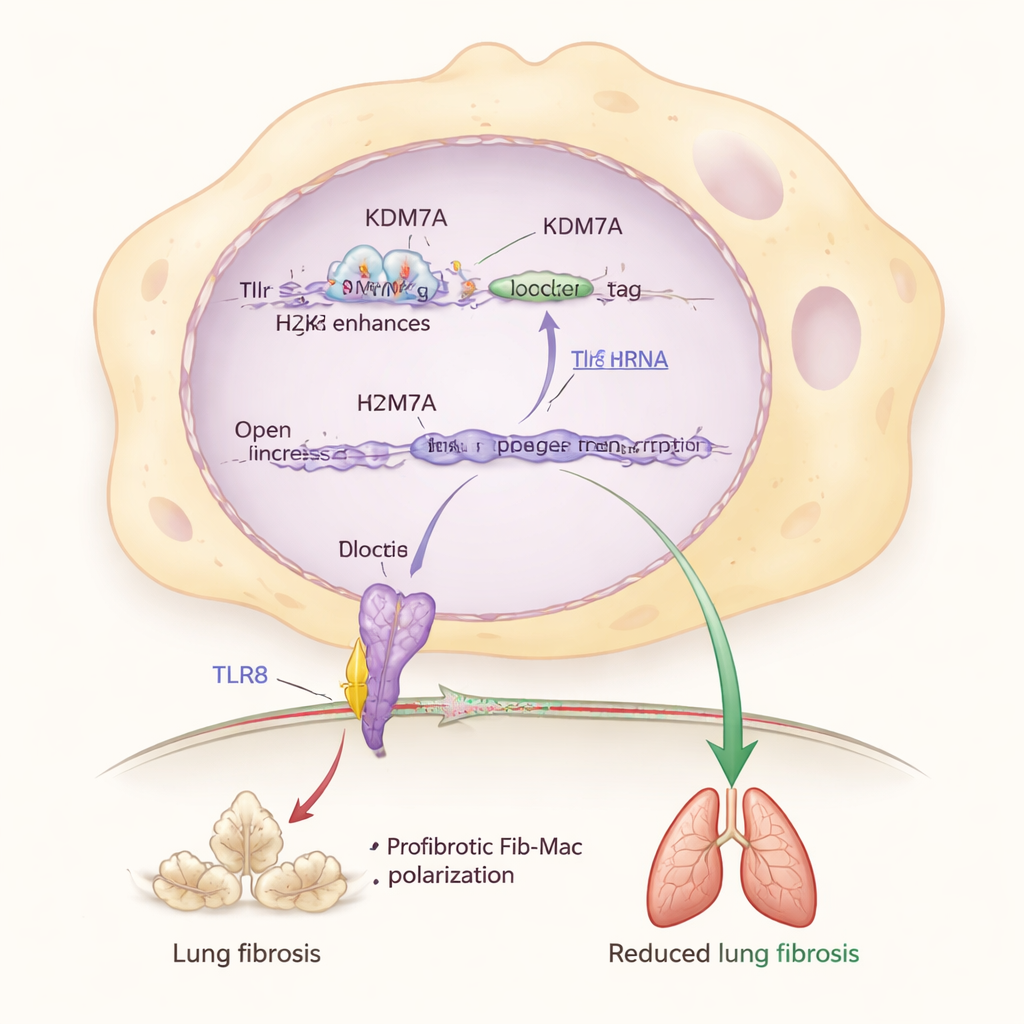
L’équipe a commencé par passer en revue des centaines de régulateurs épigénétiques connus — des protéines qui ajustent la manière dont l’ADN est emballé et quels gènes sont activés ou désactivés. À l’aide du séquençage ARN sur des macrophages humains et murins, ils ont constaté qu’une enzyme appelée KDM7A s’activait fortement lorsque les macrophages étaient orientés vers un état profibrotique et réparateur. KDM7A est une « déméthylase des histones » : elle retire certaines marques chimiques des protéines histones autour desquelles l’ADN est enroulé. Ce profil suggérait que KDM7A pourrait agir comme un frein de rétroaction intégré, activé précisément quand les macrophages commencent à dériver vers une identité favorisant la cicatrisation.
Pour tester cette hypothèse, les chercheurs ont utilisé des souris dépourvues du gène Kdm7a et ont provoqué une lésion pulmonaire avec le médicament de chimiothérapie bléomycine, un modèle standard de fibrose pulmonaire. Peu après la blessure, le tissu pulmonaire semblait similaire chez les animaux normaux et ceux déficients en Kdm7a. Mais au bout de trois semaines, les souris privées de Kdm7a présentaient des cicatrices beaucoup plus étendues, un effondrement des alvéoles et des scores d’Ashcroft plus élevés quantifiant la fibrose. Les gènes impliqués dans la production de collagène et d’autres voies associées à la fibrose étaient plus actifs chez ces souris knockout, confirmant que la perte de Kdm7a rend les poumons plus vulnérables à une formation de cicatrices durable et délétère.
Comment KDM7A éloigne les macrophages d’un destin pro-cicatriciel
En utilisant le séquençage ARN unicellulaire, les auteurs se sont concentrés sur les cellules pulmonaires individuelles issues de souris lésées. Ils ont découvert qu’en l’absence de Kdm7a, un sous-ensemble particulier de macrophages du tissu de soutien pulmonaire s’était considérablement élargi et avait acquis une forte signature Fib-Mac, exprimant des gènes tels que Arg1, Spp1 et Trem2. Des expériences complémentaires sur des macrophages en culture ont montré que la suppression de Kdm7a augmentait les gènes marqueurs Fib-Mac et réorientait le métabolisme cellulaire vers des voies favorisant la production de collagène et une activation soutenue. En d’autres termes, KDM7A freine normalement à la fois les programmes génétiques et métaboliques qui poussent les macrophages vers un état pro-fibrotique.
En approfondissant, les chercheurs ont identifié un partenaire clé de ce système de freinage : une protéine senseur appelée TLR8, qui détecte des fragments d’ARN à l’intérieur des cellules immunitaires. Ils ont montré que KDM7A contribue à maintenir le gène Tlr8 activé en retirant une marque chimique répressive (H3K27me2) d’une région amplificatrice proche de Tlr8. Lorsque Kdm7a était inactivé, cette marque s’accumulait, les niveaux de Tlr8 chutaient et les caractéristiques Fib-Mac s’intensifiaient. Réduire directement Tlr8 dans les macrophages les poussait aussi vers une identité fibrotique, tandis qu’activer ou surexprimer TLR8 les ramenait en arrière, même en l’absence de Kdm7a. Cela place la voie KDM7A–TLR8 au centre d’un circuit moléculaire qui protège les poumons d’une cicatrisation excessive.
Des poumons vieillissants à la maladie humaine
Pour relier ces résultats aux humains, l’équipe a examiné des tissus pulmonaires de patients atteints de maladie pulmonaire fibrotique. Comparés aux tissus témoins non malades, les poumons fibrosés contenaient beaucoup plus de macrophages portant des marqueurs Fib-Mac, mais ces mêmes cellules montraient des niveaux nettement réduits de KDM7A et de TLR8. Une réanalyse de jeux de données unicellulaires existants de patients atteints de fibrose pulmonaire idiopathique a confirmé ce schéma : à mesure que les signatures Fib-Mac augmentaient, l’expression de KDM7A diminuait. Les chercheurs ont aussi exploré une grande atlas murin et constaté que l’expression de Kdm7a et Tlr8 dans les macrophages diminuait avec l’âge chez les mâles, reflétant le risque plus élevé de fibrose pulmonaire chez les hommes âgés. Cela suggère que l’affaiblissement lié à l’âge et au sexe du frein KDM7A–TLR8 pourrait contribuer à expliquer qui est le plus vulnérable à une cicatrisation pulmonaire sévère.
Ce que cela signifie pour les traitements futurs
En termes simples, ce travail montre que notre système immunitaire porte un mécanisme de sécurité interne qui empêche les cellules de réparation utiles de devenir excessives et de se transformer en moteurs de cicatrices permanentes. KDM7A, agissant via TLR8, empêche les macrophages de se verrouiller dans un mode profibrotique et aide ainsi à préserver un tissu pulmonaire souple et fonctionnel après une lésion. Lorsque ce système faiblit — par perte génétique, vieillissement ou autres facteurs — les macrophages sont plus susceptibles de devenir des « amplificateurs de cicatrices », aggravant la fibrose. En révélant ce frein épigénétique, l’étude oriente vers de nouvelles stratégies thérapeutiques : des médicaments qui augmentent l’activité de KDM7A, en imitent les effets, ou stimulent prudemment TLR8 pourraient un jour compléter les thérapies antifibrotiques existantes et offrir une meilleure protection contre la cicatrisation pulmonaire progressive et potentiellement mortelle.
Citation: Funagura, N., Koga, T., Etoh, K. et al. Histone demethylase KDM7A negatively regulates fibrotic macrophage polarization and lung fibrosis progression. Commun Biol 9, 309 (2026). https://doi.org/10.1038/s42003-026-09610-1
Mots-clés: fibrose pulmonaire, macrophages, épigénétique, KDM7A, TLR8