Clear Sky Science · fr
Tests cellulaires et isoformes-spécifiques des kinases régulant les récepteurs couplés aux protéines G pour une évaluation complète des inhibiteurs
Pourquoi il est important d’« baisser le volume » cellulaire
Beaucoup de nos médicaments agissent en augmentant ou en diminuant l’activité de récepteurs à la surface des cellules qui détectent hormones, neurotransmetteurs et médicaments. Ces récepteurs doivent être soigneusement éteints ensuite pour éviter une surexcitation cellulaire, un processus partiellement contrôlé par des enzymes appelées GRK. Lorsque les GRK sont trop actives, comme cela se produit dans l’insuffisance cardiaque et certains cancers, la signalisation devient déréglée. Cette étude développe des tests pratiques et cellulaires pour mesurer dans quelle mesure des molécules expérimentales peuvent bloquer des GRK spécifiques, aidant les chercheurs à concevoir des médicaments plus intelligents qui ajustent finement ces boutons de volume cellulaires cruciaux.
Des gardiens à la surface des cellules
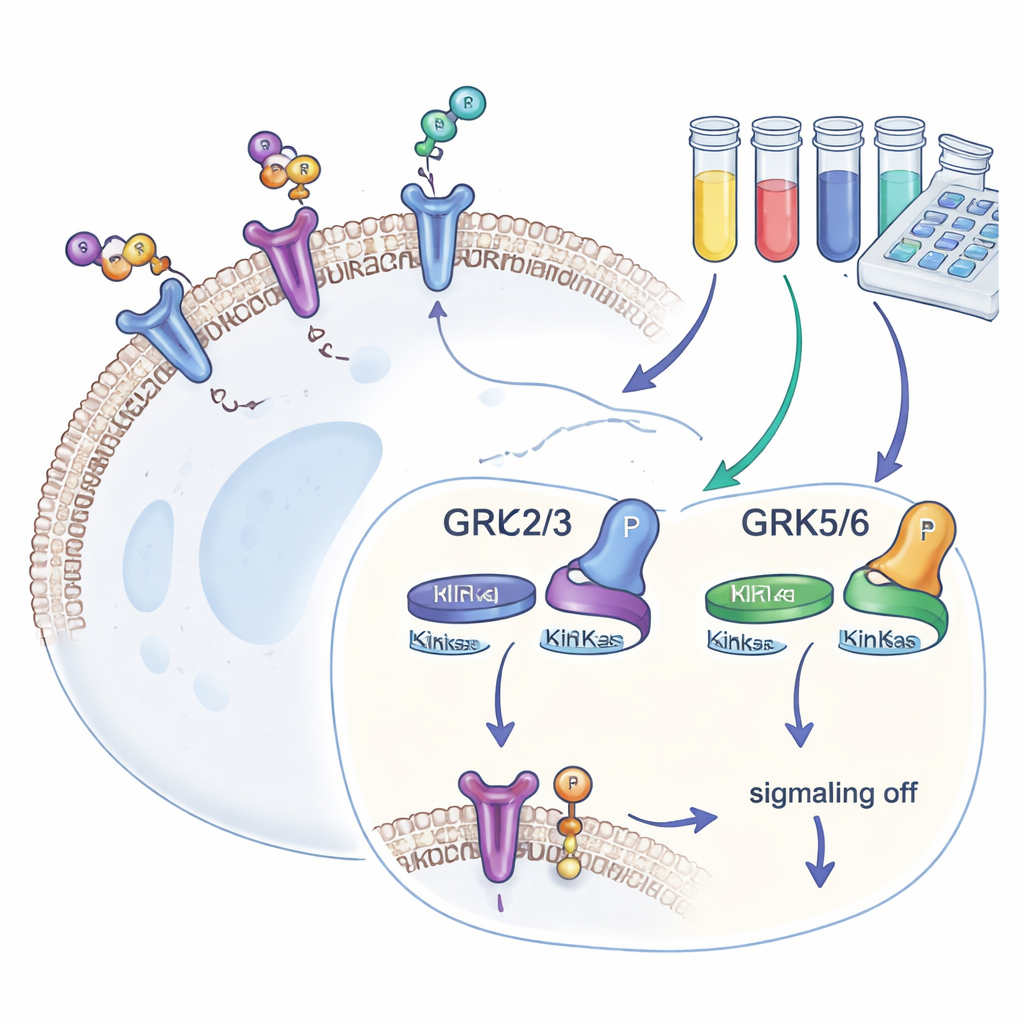
Nos cellules possèdent des centaines de types de récepteurs couplés aux protéines G (GPCR), qui détectent les signaux externes et les transduisent en réponses internes. Après l’activation d’un GPCR, les GRK fixent de petites « marques » phosphate sur sa queue. Ces marques attirent une autre protéine, la bêta-arrestine, qui stoppe la signalisation supplémentaire et entraîne souvent l’internalisation du récepteur. Quatre isoformes de GRK — GRK2, GRK3, GRK5 et GRK6 — sont présentes dans de nombreux tissus. Parce qu’elles modulent l’intensité de la réponse des GPCR et que leurs niveaux sont altérés dans des maladies comme l’insuffisance cardiaque, le cancer et l’addiction, les développeurs de médicaments cherchent des bloqueurs de GRK à la fois puissants et sélectifs.
Construire un banc d’essai propre à l’intérieur des cellules
La plupart des études antérieures sur les GRK reposaient sur la modélisation informatique ou la chimie en éprouvette, qui révèlent l’affinité d’un inhibiteur mais pas sa performance dans l’environnement encombré d’une cellule vivante. Pour combler cette lacune, les auteurs ont modifié des cellules humaines HEK293 dépourvues des quatre GRK courants, puis ont réintroduit une seule isoforme de GRK à la fois. Chaque lignée cellulaire portait aussi un récepteur bien étudié, le récepteur bêta-2 adrénergique, marqué de sorte que sa phosphorylation sur un site précis de la queue (appelé T360/S364) puisse être lue à l’aide d’un test sensible basé sur des anticorps. Comme ce site est modifié uniquement par les GRK, la quantité de phosphate présente sert de mesure directe et quantitative de l’activité de chaque isoforme de GRK dans des cellules vivantes.
Classer les bons, les faibles et les non spécifiques
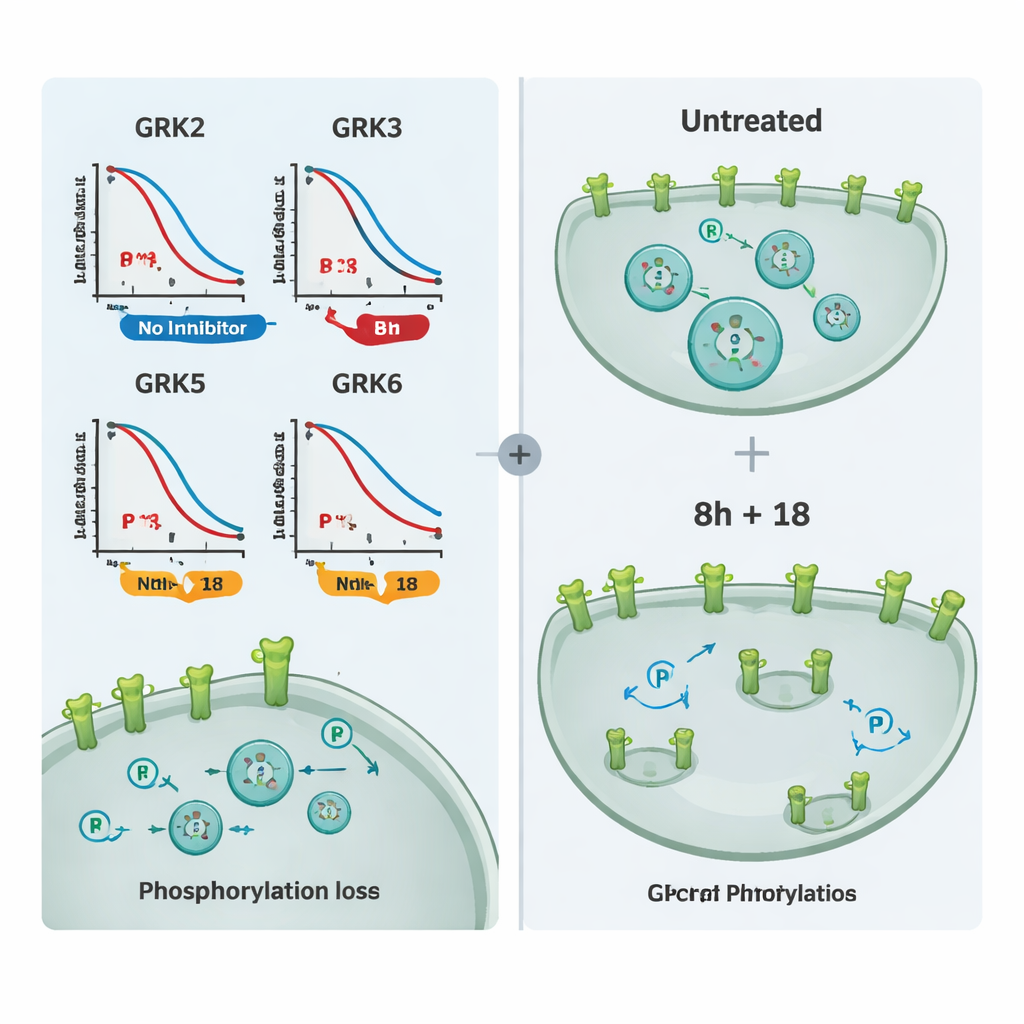
Avec cette boîte à outils, l’équipe a testé un panel d’inhibiteurs de GRK disponibles commercialement. Ils ont d’abord regroupé des composés ciblant principalement GRK2 et GRK3, et un autre ensemble visant GRK5 et GRK6. En comparant dans quelle mesure chaque molécule réduisait la phosphorylation du récepteur dans des cellules exprimant une seule sous-unité de GRK, ils ont pu cartographier la sélectivité en conditions réelles. Un composé, appelé 8h, est apparu comme le bloqueur le plus puissant de GRK2/3, tandis que le composé 18 s’est distingué par son inhibition sélective de GRK5/6. Certains molécules largement utilisées ont montré peu d’effet aux doses testées, probablement parce qu’elles n’entraient pas efficacement dans les cellules, et un inhibiteur covalent très puissant perturbait la santé cellulaire, le rendant inadapté aux expériences d’imagerie.
Des empreintes chimiques au comportement des récepteurs
Pour montrer que ces inhibiteurs affectent non seulement un récepteur-test mais la biologie des GPCR plus largement, les auteurs ont examiné plusieurs récepteurs d’intérêt médical, y compris le récepteur mu-opioïde et le récepteur de la vasopressine V2. Ils ont mesuré à la fois la phosphorylation et l’internalisation des récepteurs par microscopie. Le composé 8h ou 18 seul a réduit partiellement la phosphorylation et le déplacement vers l’intérieur des récepteurs pour de nombreuses cibles, mais la combinaison de 8h et 18 a presque complètement empêché ces changements et a maintenu les récepteurs à la surface cellulaire. Des expériences supplémentaires suivant le recrutement de la bêta-arrestine ont confirmé que ces mêmes composés pouvaient ajuster la signalisation d’autres récepteurs régulés par des ensembles de GRK qui se chevauchent.
Ce que cela signifie pour les médicaments à venir
Pour les non-spécialistes, le message clé est que l’étude fournit un jeu fiable de tests cellulaires — et deux composés-outils particulièrement utiles, 8h et 18 — qui permettent aux chercheurs de voir, dans des cellules vivantes, exactement comment les différentes isoformes de GRK sont atténuées. Plutôt que de deviner à partir de données simplifiées en éprouvette, les scientifiques peuvent désormais comparer côte à côte des inhibiteurs candidats et décider s’ils affectent surtout GRK2/3, GRK5/6, ou les quatre à la fois. Cette clarté devrait accélérer le développement de médicaments qui moduleraient plus précisément la signalisation des GPCR, avec des bénéfices potentiels pour le traitement des maladies cardiaques, du cancer, des troubles de la douleur et d’autres affections où l’équilibre de la signalisation est perturbé.
Citation: Blum, N.K., Kiefer, M.C., Decker, A. et al. Cell-based and isoform-selective G protein-coupled receptor kinase assays for comprehensive inhibitor evaluation. Commun Biol 9, 287 (2026). https://doi.org/10.1038/s42003-026-09568-0
Mots-clés: Signalisation des GPCR, Inhibiteurs de GRK, Récepteur bêta-adrénergique, essai cellulaire, découverte de médicaments