Clear Sky Science · fr
Améliorer la prédiction de kcat via un mécanisme d’attention sensible aux résidus et des représentations pré-entraînées
Pourquoi des prédictions enzymatiques plus rapides comptent
Les enzymes sont les petites machines qui maintiennent les cellules — et des industries entières — en fonctionnement. Elles accélèrent les réactions chimiques qui alimentent notre métabolisme, fabriquent des médicaments et favorisent des procédés de production plus écologiques. Un paramètre clé décrivant la vitesse d’une enzyme est le nombre de turnover, ou kcat. Mesurer le kcat en laboratoire est long et coûteux, si bien que les scientifiques se tournent vers l’intelligence artificielle pour le prédire à partir de la séquence et des informations sur la réaction. Cet article présente PMAK, un nouveau modèle d’IA qui non seulement prédit le kcat plus précisément que les outils antérieurs, mais aide aussi à identifier quelles parties d’une enzyme sont les plus importantes pour son activité.
Du travail laborieux au laboratoire aux prédictions intelligentes
Traditionnellement, déterminer le kcat implique de mesurer soigneusement la vitesse à laquelle une enzyme convertit son substrat en produit dans des conditions strictement contrôlées, comme la température et le pH. Faire cela pour des milliers d’enzymes est impraticable, ce qui limite notre capacité à modéliser des réseaux métaboliques entiers ou à concevoir de nouveaux biocatalyseurs. Les méthodes informatiques antérieures ont tenté de combler cette lacune, mais beaucoup reposaient sur des caractéristiques conçues manuellement ou sur une vision simplifiée de l’enzyme et d’un unique substrat. Elles fonctionnaient souvent bien uniquement lorsque les nouvelles enzymes étaient très similaires à celles présentes dans les données d’entraînement, et elles peinaient avec des enzymes véritablement nouvelles, des réactions inédites ou des mutants modifiés.
Apprendre aux ordinateurs le « langage » des enzymes et des réactions
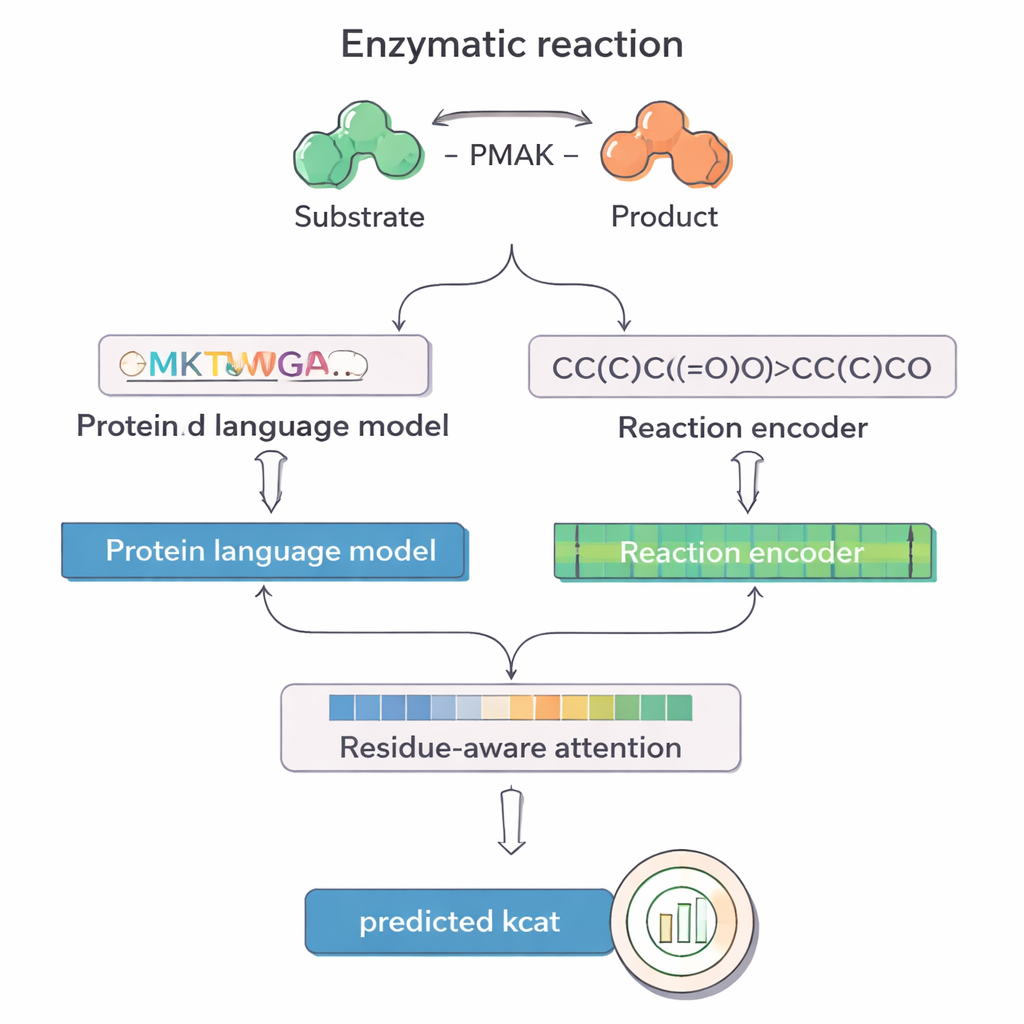
PMAK tire parti des progrès récents des « modèles de langage » développés à l’origine pour le texte, mais réentraînés sur d’immenses collections de séquences protéiques et de réactions chimiques. Un modèle, nommé ProT5, transforme la séquence d’acides aminés d’une enzyme en une représentation numérique riche qui capture des motifs appris à partir de millions de protéines. Un autre modèle, RXNFP, fait de même pour des réactions entières écrites en SMILES, qui encodent tous les réactifs et produits. PMAK alimente ces deux représentations apprises dans un réseau de neurones qui aligne leurs dimensions et permet au modèle de considérer conjointement l’enzyme et le contexte complet de la réaction, plutôt que de les traiter séparément.
Mettre en lumière les blocs de construction les plus importants
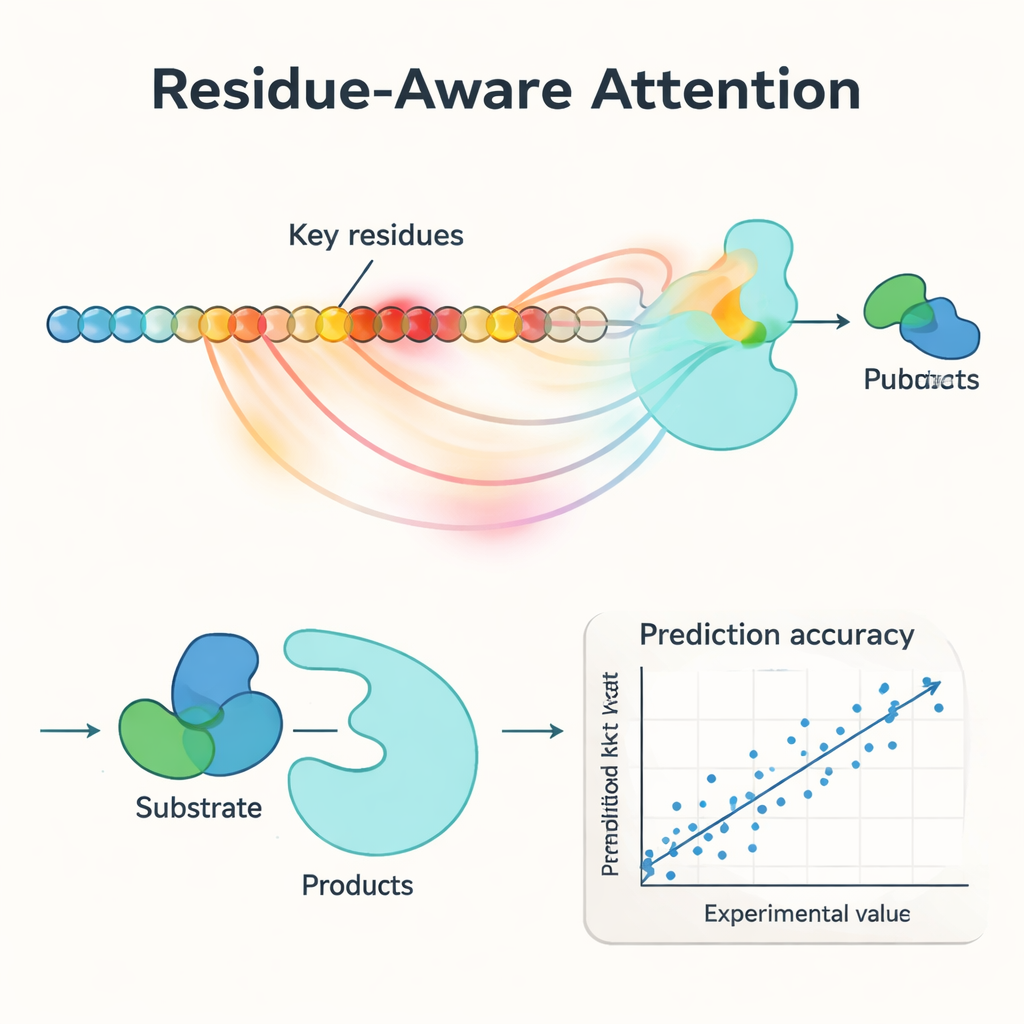
Une innovation centrale de PMAK est un mécanisme d’« attention sensible aux résidus ». Plutôt que de traiter chaque acide aminé d’une enzyme comme également important, le modèle apprend à attribuer des poids plus élevés à des résidus spécifiques qui comptent le plus pour la réaction considérée. Ces scores d’attention jouent comme un projecteur sur la séquence : lorsque les chercheurs les ont comparés aux sites actifs et de liaison connus à partir de structures protéiques, ils ont constaté que PMAK mettait systématiquement en avant des résidus fonctionnels bien plus souvent que par hasard. Le modèle a également bien fonctionné lorsque les sites actifs étaient définis plus largement pour inclure les résidus voisins dans l’espace 3D, ce qui suggère qu’il capture des indices structurels et chimiques subtils pertinents pour la catalyse.
De bonnes performances sur des enzymes nouvelles, des réactions inédites et des mutants
Les auteurs ont testé de manière rigoureuse PMAK sur un jeu de données soigné de plus de 4 000 valeurs de kcat couvrant près de 3 000 enzymes et 2 800 réactions. Dans des conditions de « warm‑start » — où des enzymes et réactions similaires apparaissent à la fois dans les ensembles d’entraînement et de test — PMAK a égalé ou surpassé les meilleurs modèles existants. Plus impressionnant encore, dans des tests « cold‑start » où soit l’enzyme soit la réaction du jeu de test n’avait jamais été vue auparavant, PMAK a surperformé une série de méthodes de pointe. Il est resté utile même pour des enzymes présentant une très faible similarité de séquence avec les données d’entraînement et pour des réactions très différentes de celles dont il avait appris. PMAK a aussi amélioré les prédictions dans des applications réalistes, comme l’estimation de la manière dont les cellules allouent leurs ressources protéiques limitées et la prévision des effets de mutations dans des jeux de données d’ingénierie enzymatique.
Ce que cela signifie pour la biologie et la biotechnologie
Pour les non‑spécialistes, PMAK peut être vu comme un assistant intelligent qui apprend à partir d’immenses « bibliothèques » de protéines et de réactions pour estimer la vitesse d’une enzyme donnée dans une réaction particulière — et pour expliquer quels acides aminés sont à l’origine de ce comportement. En combinant une précision renforcée et une compréhension au niveau des résidus, cette approche peut aider les chercheurs à concevoir de meilleures enzymes, construire des modèles métaboliques plus fiables et explorer comment les mutations affectent la fonction sans réaliser chaque expérience en laboratoire. À mesure que des modèles similaires s’étendront à d’autres traits cinétiques, ils pourraient devenir des outils essentiels pour concevoir des procédés industriels plus propres, optimiser des micro-organismes pour une production durable et approfondir notre compréhension de la façon dont les machines moléculaires de la vie atteignent leur vitesse remarquable.
Citation: Cai, Y., Ge, F., Zhang, C. et al. Enhancing kcat prediction through residue-aware attention mechanism and pre-trained representations. Commun Biol 9, 273 (2026). https://doi.org/10.1038/s42003-026-09551-9
Mots-clés: cinétique enzymatique, apprentissage profond, prédiction de kcat, ingénierie des protéines, modélisation métabolique