Clear Sky Science · fr
haCCA : intégration multi-modules des transcriptomes et métabolomes spatiaux basés sur des spots
Pourquoi cartographier les molécules in situ importe
Nos organismes sont organisés en d’innombrables petits quartiers cellulaires, chacun avec sa propre combinaison de gènes actifs et de substances chimiques. Jusqu’à récemment, les scientifiques devaient étudier ces molécules après avoir broyé les tissus en une pâte homogène, perdant toute information sur le « où ». Cet article présente une nouvelle méthode computationnelle, appelée haCCA, qui assemble deux techniques d’imagerie puissantes pour permettre aux chercheurs de voir, in situ, comment les gènes et les petites molécules se répartissent à travers des tissus et des tumeurs réels. Ce type de carte peut révéler des motifs de maladie cachés et suggérer des traitements plus précis.
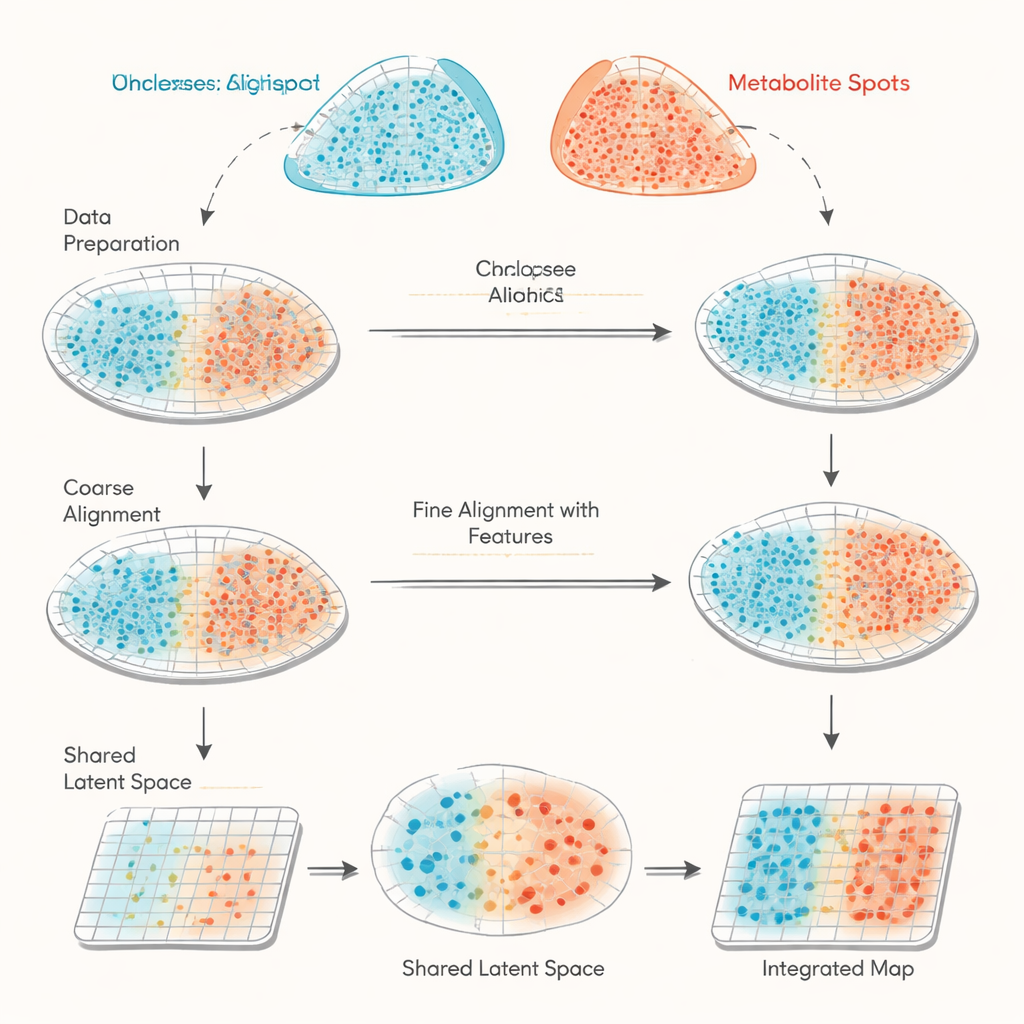
Deux regards différents sur le même tissu
L’étude porte sur la combinaison de données provenant de deux méthodes spatiales de plus en plus utilisées en biologie. La transcriptomique spatiale enregistre quels gènes sont exprimés à des milliers de petits points répartis sur une coupe de tissu. L’imagerie par spectrométrie de masse MALDI mesure les quantités de nombreuses petites molécules, comme les métabolites et les lipides, sur des grilles de points d’une densité comparable. Le problème est que ces deux instruments ne mesurent pas les mêmes positions exactes ni le même ensemble de caractéristiques, de sorte que leurs données ressemblent à deux cartes mal alignées avec des légendes différentes. Les approches existantes tentent principalement d’aligner les formes des sections tissulaires sur la base de leurs coordonnées seules, ce qui peut manquer de précision et ne fournit pas de moyen de vérifier réellement la qualité de l’alignement.
Une façon plus intelligente d’aligner les cartes moléculaires
haCCA (abréviation de hierarchical anchor-guided canonical correlation analysis) relève ce défi en combinant la géométrie et la biologie. D’abord, il effectue un « alignement morphologique » en deux étapes des grilles de spots issues des deux technologies. Des experts humains choisissent quelques repères correspondants sur les images tissulaires pour corriger grossièrement les translations et rotations, puis une étape automatisée affine les éléments aberrants près des bords déchirés ou des zones manquantes. Ensuite, la méthode identifie des paires de spots « ancrés » qui sont proches dans l’espace et situés dans des régions localement homogènes, ce qui les rend susceptibles de représenter la même zone tissulaire. À partir de ces spots ancrés, haCCA calcule quels gènes et métabolites ont tendance à varier ensemble et les condense en une représentation partagée de faible dimension capturant leurs motifs conjoints les plus forts.
Transformer les corrélations en une image tissulaire unifiée
Avec à la fois les coordonnées spatiales et la représentation moléculaire partagée en main, haCCA résout un problème d’optimisation pour estimer la probabilité d’appariement de chaque spot de gènes avec chaque spot de métabolites. Cette étape est conçue pour garder les spots proches spatialement tout en étant similaires dans leur profil combiné gène–métabolite. Le résultat final est un « plan de transport » qui relie chaque point d’un jeu de données à son partenaire optimal dans l’autre, produisant une carte multimodale intégrée. Sur des jeux de test soigneusement construits — où les relations vraies sont connues — les auteurs montrent que chaque étape du flux de travail (alignement grossier, alignement affiné et appariement tenant compte des caractéristiques) améliore progressivement trois mesures indépendantes de précision. Comparé à d’autres outils qui s’appuient principalement sur la géométrie, haCCA obtient systématiquement un meilleur alignement et un transfert plus fidèle des étiquettes de région.
Révéler une biologie cachée dans le cerveau et le cancer du foie
Les auteurs appliquent ensuite haCCA à de vrais tissus de cerveau de souris et de tumeurs hépatiques. Pour le cerveau, ils intègrent des données commerciales de transcriptomique spatiale avec des images de métabolites provenant des mêmes sections ou de sections voisines. La méthode préserve des territoires métaboliques connus et reconstruit des recouvrements attendus, comme la colocalisation de la dopamine avec le gène codant son enzyme clé. En regroupant conjointement gènes et métabolites, ils montrent que les données combinées distinguent des sous-régions tissulaires plus nuancées que chaque modalité seule. Dans un modèle préclinique de cholangiocarcinome intra-hépatique, un type de cancer du foie, ils utilisent haCCA pour comparer des tumeurs capables ou non de former des pièges extracellulaires de neutrophiles — des structures en réseau libérées par les cellules immunitaires. Les cartes intégrées révèlent que, lorsque ces pièges sont présents, un gène appelé Scd1 et les acides gras qui lui sont associés sont enrichis dans les régions malignes, indiquant une bascule vers un métabolisme lipidique altéré dans la tumeur.
Ce que cela signifie pour la recherche future
En termes simples, haCCA revient à aligner des photos aériennes prises avec différents appareils — l’un sensible aux contours des bâtiments, l’autre aux signatures thermiques — pour obtenir une image plus nette de ce qui se passe dans chaque pâté de maisons. En fusionnant avec précision l’emplacement des gènes actifs et l’accumulation des métabolites clés, ce flux de travail aide les scientifiques à profiler simultanément les deux aspects du comportement cellulaire : les instructions et la chimie qui en résulte. L’approche améliore les méthodes d’alignement antérieures, est fournie sous la forme d’un outil Python accessible et peut être étendue à d’autres technologies spatiales. À mesure que de telles cartes intégrées deviennent plus courantes, elles pourraient approfondir notre compréhension de la façon dont les tumeurs et d’autres tissus organisent leur métabolisme, répondent aux traitements et évoluent dans le temps.
Citation: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Mots-clés: multi-omiques spatiales, transcriptomique, métabolomique, métabolisme tumoral, intégration de données