Clear Sky Science · fr
Détermination ab initio des stabilités de phase des solides dynamiquement désordonnés : désordre rotationnel C2 dans Li2C2
Pourquoi ce solide qui change compte
De nombreuses technologies modernes reposent sur des solides capables de modifier discrètement leur structure interne lorsqu’on les chauffe ou les comprime. Ces changements, appelés transitions de phase, sont essentiels à des concepts tels que le refroidissement solide et des batteries plus sûres. Cette étude porte sur un composé simple, le carbure de lithium (Li2C2), qui passe d’une forme bien ordonnée à une forme plus agitée et dynamiquement désordonnée lorsque la température augmente. En observant cette transformation atome par atome dans des simulations informatiques, les auteurs montrent comment l’« agitation » interne de petits motifs moléculaires peut faire pencher la balance entre deux structures cristallines.
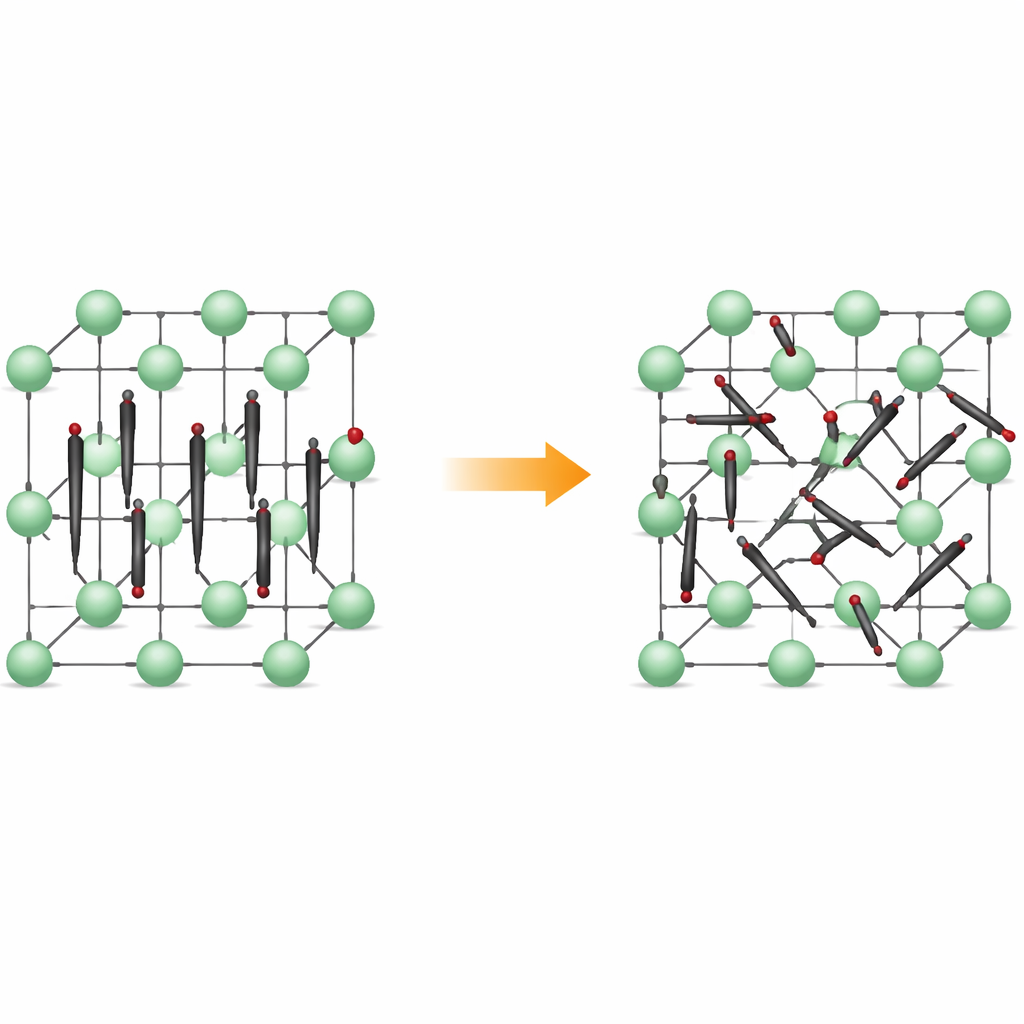
De rangées ordonnées à un mouvement incessant
À basse température, Li2C2 forme un cristal orthorhombique : ses atomes de carbone se regroupent en petits dimères C2 qui pointent presque tous dans la même direction, comme des allumettes alignées. Des ions lithium se logent entre eux, créant une trame régulière en trois dimensions. En chauffant le matériau, il se transforme en une forme cubique, où les centres des dimères restent ordonnés sur un réseau, mais où les dimères eux-mêmes ne conservent plus une direction fixe. Ils tournent plutôt entre plusieurs orientations préférentielles, passant du temps dans des vallées d’énergie peu profondes correspondant à des alignements spécifiques. Le matériau reste solide, mais sa structure interne devient dynamiquement désordonnée.
Suivre le changement le long d’un trajet continu
Pour savoir quelle phase est la plus stable à une température donnée, il faut comparer leurs énergies libres, qui combinent énergie et entropie (une mesure du désordre). Les méthodes standard basées sur de petites vibrations autour de positions fixes peinent lorsque les atomes se déplacent ou tournent de manière significative. Ici, les auteurs utilisent une technique appelée intégration thermodynamique contrainte–déformation, fondée sur la dynamique moléculaire de premiers principes. Ils construisent un trajet de déformation continu qui transforme progressivement la cellule de simulation de la structure orthorhombique basse température vers la structure cubique haute température. Le long de ce trajet, ils effectuent de longues simulations à température constante et mesurent la réponse des contraintes internes à la déformation imposée. L’intégration de cette réponse en contrainte donne la différence d’énergie libre entre les deux phases.
Voir l’entropie à travers le mouvement atomique
Les calculs révèlent qu’autour de 600 K la phase orthorhombique basse température est encore légèrement favorisée, tandis qu’à 650 K la phase cubique l’emporte par quelques millièmes d’électron-volt par unité de formule. L’interpolation entre ces résultats donne une température de transition d’environ 611 K. Cela est inférieur aux estimations expérimentales mais reste en accord raisonnable, compte tenu des faibles différences d’énergie libre en jeu. L’énergie interne de la phase cubique est en réalité plus élevée ; ce qui la stabilise est un important gain d’entropie, directement attribué au désordre rotationnel des dimères C2. En analysant la façon dont l’orientation de chaque dimère perd la mémoire de sa direction initiale au fil du temps, les auteurs montrent que les dimères se réorientent à l’échelle du sous-picoseconde, estompant la frontière entre les catégories habituelles d’entropie « vibrationnelle » et « configurationnelle ».
Au-delà des images simplistes du désordre solide
Le travail souligne également que les raccourcis courants — comme traiter l’entropie comme la somme simple des vibrations autour de configurations fixes plus un comptage séparé des orientations statiques — échouent pour des matériaux comme Li2C2. Parce que les rotations de dimères sont rapides et fortement couplées aux vibrations ordinaires, le système ne peut pas être proprement décomposé en parties « vibrant » et « se réarrangeant » séparées. La méthode d’intégration contrainte–déformation contourne cette difficulté : elle extrait l’énergie libre complète directement à partir de la dynamique microscopique, sans avoir à deviner comment l’entropie devrait être répartie.
Ce que nous apprend l’étude
En termes simples, l’étude montre comment un solide peut rester rigide tandis que ses éléments constitutifs internes deviennent de plus en plus libres de pivoter et de tourner, et comment cette liberté interne peut rendre une structure plus désordonnée préférée thermodynamiquement. Pour Li2C2, la phase cubique haute température est stabilisée non pas parce qu’elle est moins coûteuse en énergie, mais parce qu’elle offre beaucoup plus de manières pour les dimères C2 de s’orienter et de bouger. En montrant que l’intégration thermodynamique contrainte–déformation peut capturer ce subtil équilibre entre ordre, énergie et entropie, ce travail ouvre la voie à la prédiction de transitions similaires dans d’autres solides dynamiquement désordonnés susceptibles de sous-tendre de futurs dispositifs de refroidissement, batteries et matériaux intelligents.
Citation: Klarbring, J., Filippov, S., Häussermann, U. et al. Ab initio determination of phase stabilities of dynamically disordered solids: rotational C2 disorder in Li2C2. Sci Rep 16, 8965 (2026). https://doi.org/10.1038/s41598-026-43795-z
Mots-clés: transition de phase en phase solide, désordre dynamique, dynamique moléculaire, carbures de lithium, intégration thermodynamique