Clear Sky Science · fr
Conception pilotée par les données d’inhibiteurs LNA pour une élimination efficace des contaminants dans les bibliothèques Ribo‑Seq
Pourquoi le nettoyage des données de séquençage est important
La biologie moderne repose souvent sur la lecture de millions de petits fragments d’ARN pour comprendre comment les cellules synthétisent des protéines. Mais ces mesures puissantes, en particulier une méthode appelée profilage des ribosomes (Ribo‑Seq), peuvent être encombrées de fragments d’ARN non pertinents qui gaspillent la puissance de séquençage et de l’argent. Cette étude décrit une manière simple et guidée par les données de concevoir des « bloqueurs » moléculaires spécialisés qui éliminent sélectivement ces fragments indésirables, doublant presque l’information utile que les chercheurs obtiennent à partir du même essai.
Le problème des instantanés de ribosomes bruités
Le Ribo‑Seq capture un instantané moment‑par‑moment des messages d’une cellule qui sont activement traduits en protéines. Pour ce faire, les scientifiques isolent les ribosomes avec les courts segments d’ARN messager (ARNm) qu’ils protègent. Tout le reste est dégradé, et les extraits protégés sont séquencés puis alignés sur le génome. En pratique, cependant, de nombreux autres petits fragments d’ARN non codants passent ce filtre. Parce que ces fragments contaminants sont abondants et fortement variables, ils accaparent une grande part des lectures de séquençage, laissant moins de lectures pour les véritables signaux codant pour des protéines qui intéressent les chercheurs.
Pourquoi les astuces de nettoyage existantes sont insuffisantes
Les stratégies standards tentent d’éliminer les ARN ribosomiques abondants et autres ARN non codants à l’aide de sondes de capture préconçues ou d’enzymes. Ces méthodes fonctionnent bien lorsque les ARN cibles sont intacts et prévisibles, mais le Ribo‑Seq fragmente volontairement l’ARN en de nombreux fragments de tailles différentes. Cette fragmentation brouille les sites ciblés par des jeux de sondes fixes, rendant la déplétion beaucoup moins efficace. De plus, le mélange exact de contaminants dépend de l’espèce étudiée, des conditions de croissance et même de l’enzyme nucléase utilisée. Les flux de travail de purification existants impliquent aussi souvent plusieurs étapes d’incubation et de purification, qui prennent du temps et peuvent provoquer des pertes d’échantillon ou des biais.
Bloqueurs personnalisés conçus à partir de données réelles
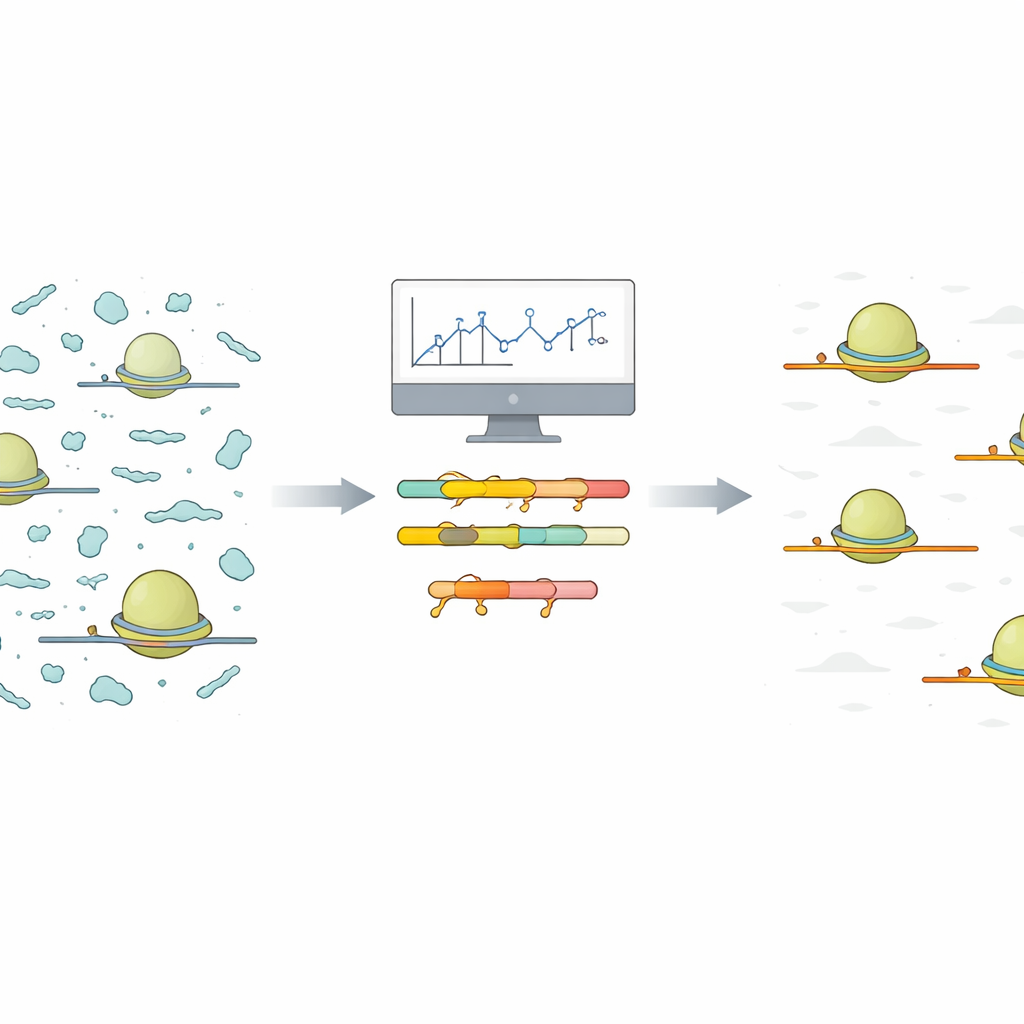
Les auteurs proposent une approche simplifiée qui commence par une petite expérience pilote de séquençage peu coûteuse, réalisée dans les mêmes conditions que l’expérience complète prévue. Ils fournissent un script R qui prend les lectures alignées de ce run pilote et groupe automatiquement les fragments contaminants similaires en fonction de leur séquence. Pour chaque groupe, le script identifie la plus courte séquence commune apparaissant dans les fragments. Ces courtes régions partagées sont des sites cibles idéaux pour des oligonucléotides appelés acides nucléiques verrouillés (LNA). Les LNA sont de courts brins portant une modification chimique qui leur permet de se lier très fermement à l’ARN complémentaire. Le script génère aussi des heatmaps et des graphiques récapitulatifs intuitifs, aidant les utilisateurs à visualiser quels contaminants dominent et combien de cibles LNA seraient nécessaires pour un nettoyage substantiel.
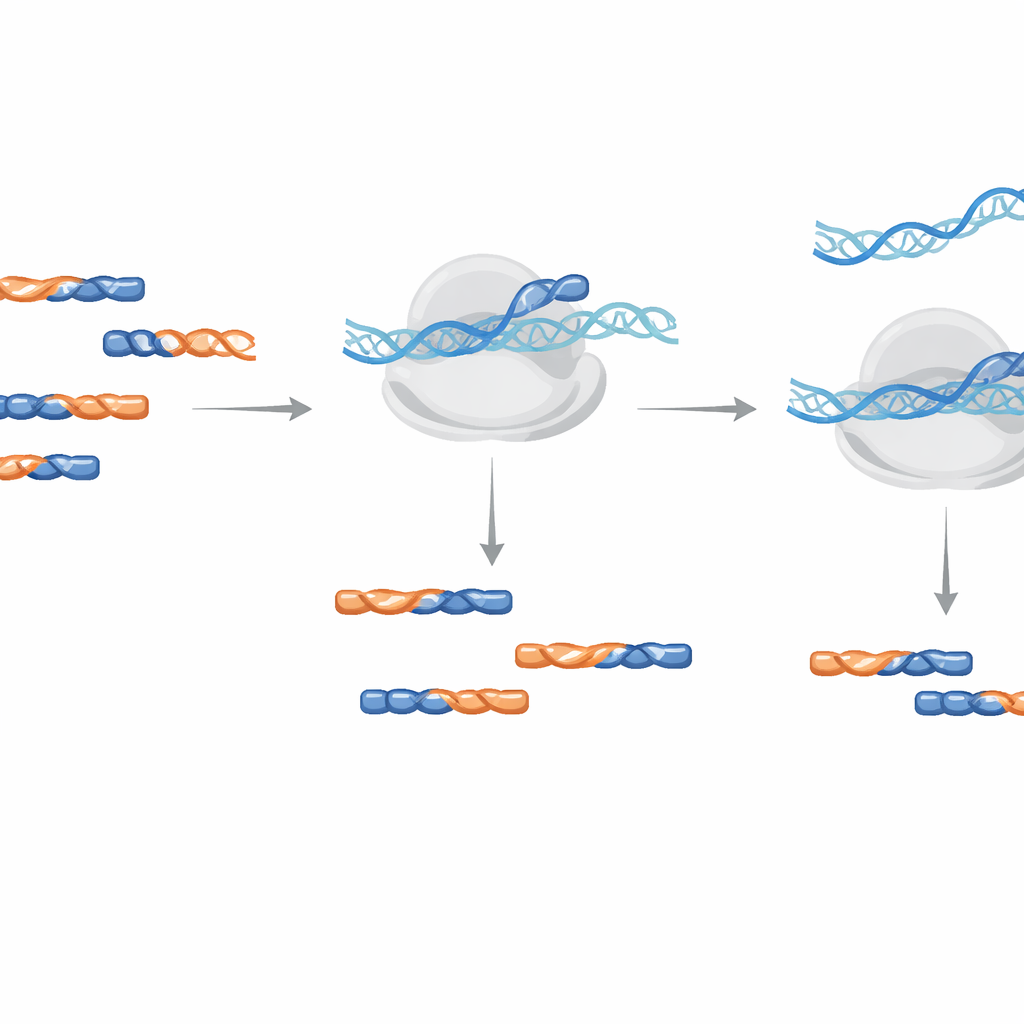
Un nettoyage en une étape pendant l’amplification
Plutôt que d’extraire physiquement les contaminants de l’échantillon, la méthode utilise des oligonucléotides LNA comme bloqueurs pendant l’étape d’amplification de l’ADN qui constitue la bibliothèque de séquençage. Les auteurs ont testé l’ajout de ces bloqueurs soit lors de la transcription inverse initiale, soit durant l’amplification PCR ultérieure. Ils ont constaté que l’ajout d’LNAs pendant l’amplification était plus efficace et nécessitait des concentrations plus faibles, réduisant un contaminant test de plus d’un millier de fois tout en fonctionnant indépendamment de l’orientation du brin. Parmi les conseils pratiques de conception figurent l’alternance de briques de construction standard en ADN et en LNA, l’utilisation d’une longueur minimale de 14 unités pour la plante Arabidopsis, et la modification de l’extrémité terminale afin que le bloqueur lui‑même ne puisse pas être étendu par erreur.
Plus de lectures utiles sans déformer le signal
Pour démontrer les performances en conditions réelles, l’équipe a conçu cinq bloqueurs LNA ciblant les groupes de contaminants les plus courants observés dans des conditions de croissance typiques chez Arabidopsis. Lorsqu’ils ont ajouté ce mélange pendant l’amplification des bibliothèques, la proportion de contaminants identifiés a chuté de plus de 30 %, et le nombre de lectures utiles codant pour des protéines a presque doublé. Fait crucial, en comparant les comptes de lectures au niveau des gènes entre bibliothèques avec et sans traitement LNA, les valeurs concordaient quasiment parfaitement, indiquant que les bloqueurs éliminaient les fragments indésirables sans déformer le signal biologique provenant des véritables empreintes d’ARNm.
Ce que cela signifie pour les expériences futures
Ce travail montre qu’une courte expérience pilote, associée à un script d’analyse facile à utiliser et à un petit jeu de bloqueurs LNA sur mesure, peut transformer des bibliothèques Ribo‑Seq encombrées en jeux de données beaucoup plus propres et informatifs en une seule étape de pipetage. Les chercheurs obtiennent plus de lectures significatives par run, réduisant les coûts et simplifiant la conception expérimentale, tout en préservant des mesures précises de la traduction des gènes. Les auteurs fournissent également des profils de contaminants prêts à l’emploi et des conceptions de bloqueurs pour des conditions végétales courantes, et suggèrent que des ressources similaires pourraient être développées pour de nombreux organismes, rendant le profilage des ribosomes de haute qualité plus accessible à la communauté de recherche.
Citation: Ricciardi, D.A., Peter, F.E. & Böhmer, M. Data-driven design of LNA-blockers for efficient contaminant removal in Ribo-Seq libraries. Sci Rep 16, 8565 (2026). https://doi.org/10.1038/s41598-026-43117-3
Mots-clés: profilage des ribosomes, contaminants ARN, acides nucléiques verrouillés, purification de bibliothèques de séquençage, régulation de la traduction