Clear Sky Science · fr
Modèle d’apprentissage profond hybride optimisé pour le diagnostic du cancer du sein à partir de données génétiques
Pourquoi cela compte pour les patients et les familles
Le cancer du sein est désormais le cancer le plus fréquemment diagnostiqué chez les femmes dans le monde, et le détecter tôt peut faire la différence entre la vie et la mort. Les médecins disposent de plus en plus souvent des informations génétiques d’un patient, mais transformer des dizaines de milliers de mesures géniques en réponses claires est extraordinairement difficile. Cet article décrit un nouveau modèle informatique qui lit ces motifs génétiques complexes pour repérer le cancer du sein et prédire les évolutions avec une précision remarquable, offrant potentiellement aux cliniciens un assistant puissant pour des décisions plus précoces et plus fiables.
Des gènes aux signaux d’alerte
Chaque tumeur du sein porte une empreinte moléculaire encodée par l’activité de milliers de gènes. Les auteurs ont voulu construire un système capable de lire directement cette empreinte, plutôt que de se fier uniquement aux images ou à une poignée de gènes bien connus comme BRCA1 et BRCA2. Ils ont travaillé avec deux des plus grandes ressources publiques en génomique du cancer : la cohorte de cancer du sein TCGA, qui comprend l’activité de 17 814 gènes dans 590 échantillons, et l’étude METABRIC, qui contient des informations génomiques et cliniques pour plus de 1 400 patientes. Leur objectif était ambitieux : concevoir une méthode capable de gérer ce flot d’informations, d’en extraire les signaux les plus révélateurs et de rester fiable sur des groupes de patients complètement indépendants.
Réduire des milliers de gènes à un ensemble utile
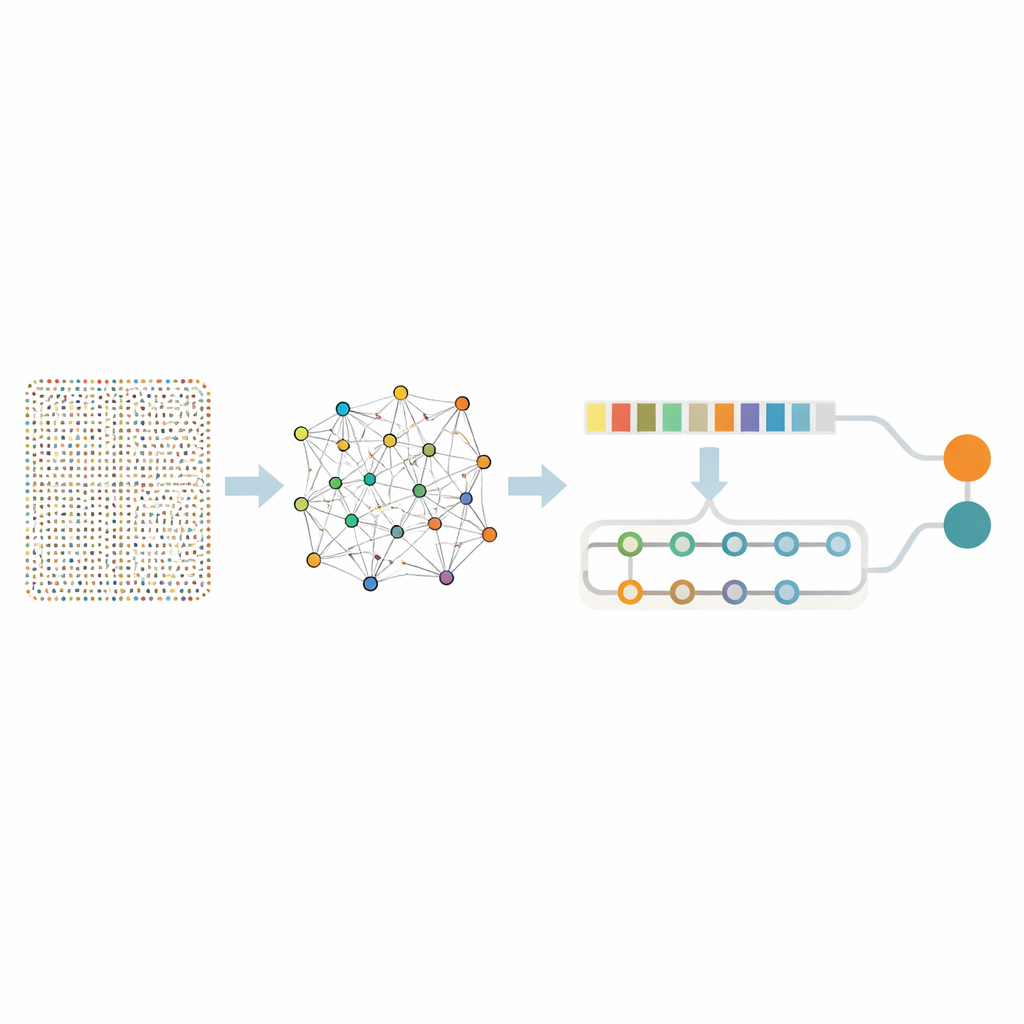
Considérer près de dix-huit mille gènes à la fois est accablant, même pour des algorithmes avancés, et cela risque de capter du bruit sans signification. Les chercheurs ont donc utilisé un « tamis » en deux étapes pour isoler un plus petit ensemble de gènes véritablement informatifs. D’abord, ils ont appliqué une technique appelée Random Forest, qui interroge en pratique de nombreux arbres de décision pour déterminer quels gènes importent le plus pour distinguer tissus tumoraux et échantillons sains. Cette étape a réduit la liste à 436 gènes prometteurs. Ensuite, ils ont examiné la façon dont ces gènes se comportent ensemble en utilisant le mining de règles d’association, une méthode qui repère des groupes de gènes qui ont tendance à être actifs simultanément dans les tumeurs. Cette couche d’analyse supplémentaire a identifié des paires et des réseaux géniques liés à des processus clés du cancer tels que la division cellulaire rapide, la réparation de l’ADN et les modifications du tissu entourant la tumeur. Après ce raffinage, 332 gènes sont restés — toujours riches en sens biologique mais beaucoup plus maniables pour une analyse approfondie.
Un réseau neuronal en deux parties qui apprend motifs et contexte
Avec cet ensemble de gènes focalisé, l’équipe a construit un modèle d’apprentissage profond hybride qui combine deux types de réseaux neuronaux. Une partie, connue comme réseau convolutionnel, balaie la liste de gènes pour capter des motifs locaux — des groupes de gènes qui ont tendance à augmenter ou diminuer ensemble. La deuxième partie, un réseau à mémoire bidirectionnelle, regarde la même information tout en suivant les relations à longue portée, capturant comment des gènes éloignés s’influencent mutuellement sur l’ensemble du profil. Avant l’entraînement, les auteurs ont équilibré les données afin que les échantillons tumoraux et non tumoraux soient représentés équitablement et ont ajouté de petites quantités de bruit artificiel, apprenant ainsi au modèle à ne pas se laisser tromper par des fluctuations aléatoires.
Performances du système dans des tests réels
Lorsque le modèle hybride a été entraîné et testé sur les données TCGA, il a distingué correctement les échantillons tumoraux des échantillons normaux avec environ 97 % de précision et une capacité quasi parfaite à séparer les deux groupes. Fait important, il a surpassé des configurations d’apprentissage profond plus simples et des outils d’apprentissage machine classiques tels que la régression logistique et les machines à vecteurs de support, même lorsque ces méthodes concurrentes recevaient les mêmes gènes soigneusement sélectionnés. Le test le plus exigeant a toutefois été de savoir si le modèle tiendrait sur un ensemble de données entièrement différent. Appliqué à METABRIC, collecté dans d’autres hôpitaux et selon des méthodes de laboratoire différentes, le système a maintenu de hautes performances : lors de sa meilleure exécution il a atteint 99,3 % de précision et a correctement identifié chaque patiente décédée par la suite d’un cancer du sein, une propriété cruciale si l’outil doit servir à signaler les cas à haut risque.
Ce que cela pourrait signifier pour les soins futurs
Pour un non-spécialiste, l’essentiel est que cette étude fournit un filtre intelligent et un lecteur de données génétiques capables de repérer le cancer du sein et les risques associés avec une constance remarquable à travers de larges cohortes. En combinant une stratégie réfléchie de sélection des gènes avec un réseau neuronal à deux branches, les auteurs démontrent que les ordinateurs peuvent extraire des signaux cliniquement significatifs d’ensembles de données génétiques énormes, non seulement dans une étude mais à travers des cohortes indépendantes. Bien que des travaux supplémentaires soient nécessaires pour tester l’approche dans des populations diversifiées et pour expliquer ses décisions en détail, la méthode ouvre la voie à un futur où un simple prélèvement sanguin ou tissulaire pourrait alimenter de tels modèles et aider les médecins à détecter les tumeurs plus tôt et à adapter les traitements avec plus de précision.
Citation: Hesham, F., Abbassy, M.M. & Abdalla, M. Hybrid tuned deep learning model for breast cancer diagnosis using genetic data. Sci Rep 16, 9664 (2026). https://doi.org/10.1038/s41598-026-41643-8
Mots-clés: génomique du cancer du sein, diagnostic par apprentissage profond, biomarqueurs d’expression génique, détection précoce du cancer, support à la décision clinique